

随着全球能源需求持续增长与环境问题日益严峻, 开发清洁、可再生且低成本的能源转换技术成为21世纪科学研究的重要议题. 在众多可再生能源中, 太阳能因其资源丰富、分布广泛的特点, 被视为最具潜力的解决方案之一. 作为新型清洁能源技术, 有机太阳能电池(Organic Solar Cells, OSCs)因其质量轻、柔性、可溶液制备和可卷对卷大面积印刷等优点, 受到科研工作者的广泛关注. 其中, 活性层中的给受体材料是决定有机光伏器件效率的关键[1-3]. 某种程度上, OSCs的发展史也是一部受体材料的创新史. 从早期的富勒烯衍生物到如今的高性能非富勒烯受体(Non-fullerene acceptors, NFAs, 图1), 受体材料的每一次革新都大幅提升了OSCs的光电转换效率(Power conversion efficiencies, PCEs)和稳定性, 并推动了其从实验室研究走向商业化应用.
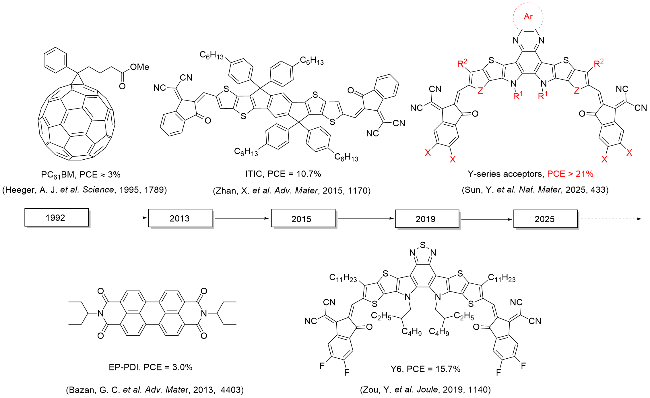
1992年, Wudl等[4]发现C60可以作为电子受体, 与共轭聚合物聚[4-亚苯基亚乙烯基](MEH-PPV)形成光诱导电荷转移, 奠定了有机光伏(OPV)的基本工作原理. 1995年, Heeger等[5]提出“体异质结(Bulk Hetero Junction, BHJ)”的概念, 将共轭聚合物MEH-PPV与可溶性富勒烯衍生物PC61BM共混, 大幅提升激子分离效率, 使OSCs器件效率接近3%. 在之后的10~15年时间里, 富勒烯衍生物因其较高的电子亲和势以及电荷传输的各向同性, 在受体材料中占主导地位[6-9]. 然而其在可见光和近红外区的吸收偏弱, 合成分离过程繁琐, 结构可修饰性差, 带隙难以调节以及溶解性差等问题, 阻碍了OSCs领域进一步发展, 目前基于富勒烯受体的光伏器件最高效率仅为11.3%[10]. 作为替代富勒烯受体的初步尝试, 苝二酰亚胺(Perylene Diimide, PDI)类小分子因其庞大的刚性π共轭体系拓宽了分子吸收[11-14], 弥补了富勒烯在可见光区吸收不足的缺点, 提升了材料的光热稳定性和载流子迁移率. 但由于未解决其相分离尺寸过大的形貌问题, 器件性能依旧没能得到根本改善. 2010年前后, 基于寡聚噻吩类[15]、苯并二噻吩类(BDT)[16]和茚并二噻吩类(IDT)[17]等A-D-A型柔性小分子给体材料的出现, 与富勒烯类受体获得了良好的能级匹配与形貌兼容, 从而帮助基于富勒烯受体的光伏器件实现了超10%的转换效率. 得益于小分子给体的设计思路, 研究人员发现这类小分子的吸收带普遍由D核的π-π*跃迁和D到A核的分子内电荷转移(Intramolecular Charge Transfer, ICT)跃迁组成. 其中封端A核的缺电子程度决定着小分子材料ICT效应的强度, 进而影响到分子的带隙和能级. 换言之, A核的缺电子特性越强, 分子的能级(尤其是LUMO能级)越低, 带隙越窄, 电荷分离的驱动力越大. 当通过增强A核的缺电子效应, 使这些低能级、窄带隙的共轭小分子与富勒烯类受体的能级与带隙接近, 甚至更低时, 便形成了NFAs. 而柔性侧链则解决了传统富勒烯和PDI类受体可溶液加工性能差的问题. 2015年, 占肖卫课题组[18]首次以对称的茚并二噻吩[3,2-b]噻吩(IDTT)为D核, 强缺电子的3-(二氰基亚甲基)茚-1-酮(IC)为封端A单元, 开发了窄带隙非富勒烯受体3,9-双(2-亚甲基-(3-(1,1-二氰基亚甲基)-茚酮))-5,5,11,11-四(4-己基苯基)-二噻吩并[3'-d']-s-茚并[6-b']二噻吩(ITIC), 实现了超800 nm吸收边的宽吸收光谱和突破10%的器件效率, 奠定了基于NFAs有机太阳能电池的基础. 羰基和丙二腈两个强吸电子官能团的引入使IC及其衍生结构, 包括2-(5,6-二氟-3-氧代-2,3-二氢-1H-茚-1-亚基)丙二腈(2F-IC)、2-(3-氧代-2,3-二氢-1H-环戊并[b]萘-1-亚基)丙二腈(NC)等均表现出强烈的缺电子特性, 对拓宽分子吸收至近红外区, 增强分子间的π-π堆积, 提升电荷迁移率起到了至关重要的作用, 也是目前大多数高效NFAs所采用的端基类型. 2019年, 邹应萍组[19]开发出吸收超过900 nm, 效率接近16%的A-DA'D-A型受体Y6, 彻底改变了OSCs领域的竞争格局. Y6一方面继承了ITIC受体原有的梯形稠环结构, 一方面打破了ITIC的C2h对称性“S”型骨架, 实现了C2v对称性“C”型骨架的结构创新, 并直接推动后续衍生物, 如L8-BO[20]、BTP-eC9[21]等高效NFAs材料的出现, 因而“C”型骨架被认为是高效NFAs的必备分子结构, 并被广泛应用于Y6衍生物(以下称作Y系列受体)的构型优化. 截至本文发表, 基于Y系列受体的小面积器件效率已经达到了令人惊叹的20%[22], 大面积器件效率达到了18%[23], 标志着OSCs商业化进程的加速.
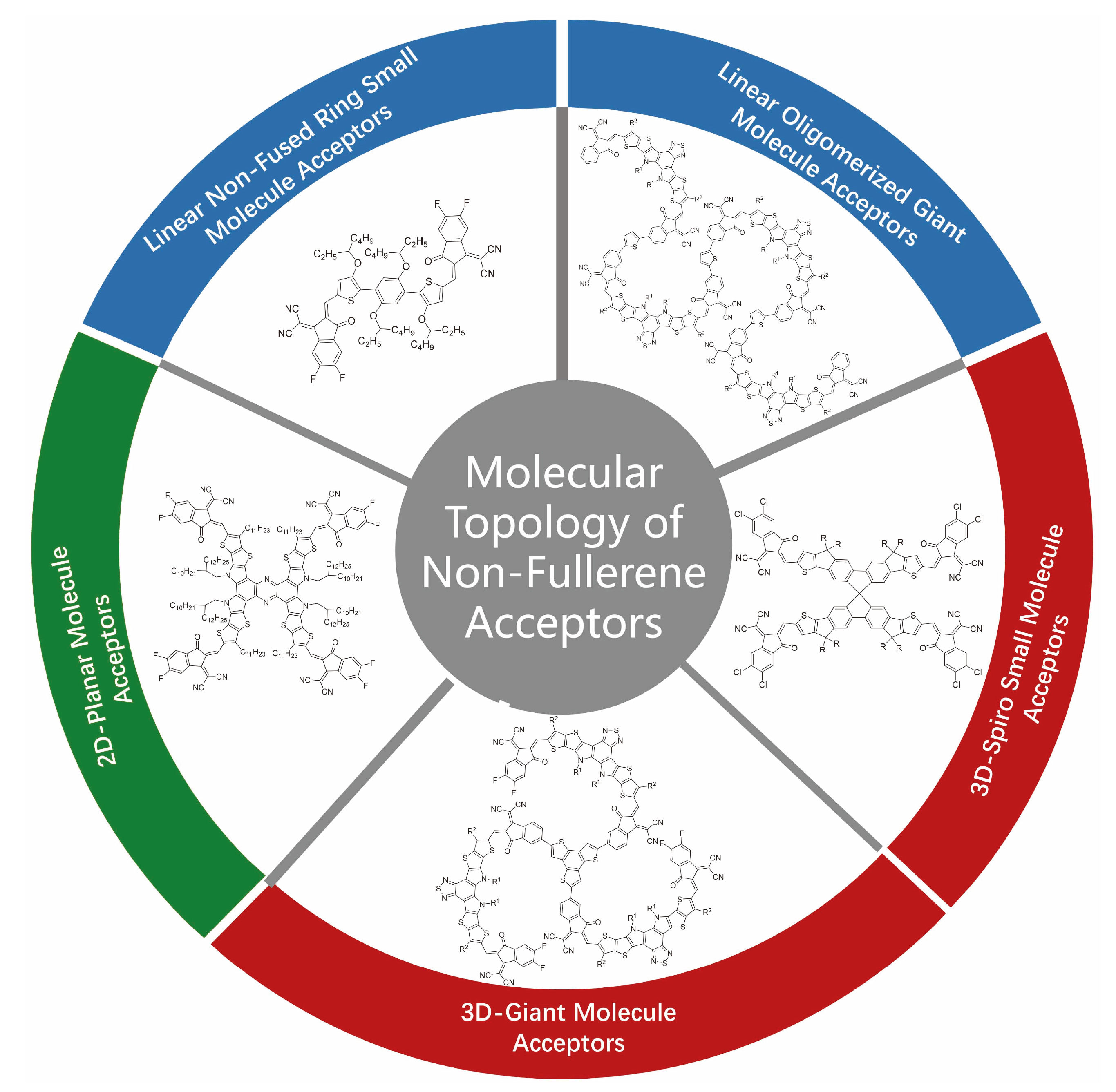
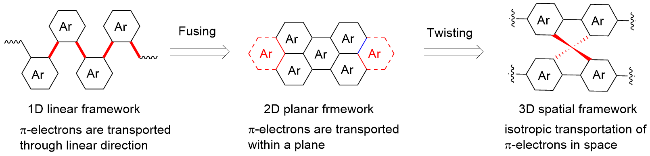
从共轭分子电子态调控调节材料特性的角度出发,对上述分子进行共轭单元、端基推拉电子单元、分子侧链单元等进行分子结构修饰, 可以有效改性材料特性, 进而开发出光电性能更为优异的非富勒烯受体材料. 包西昌[24]以及He[25]等分别从Y系列受体的改性修饰, 对该类分子的研究进展进行了综合论述. 除此之外, 詹传郎等从分子硒吩工程的角度, 对NFAs的分子结构设计进行了相关的综述[26]. 以上这些综述对Y系列分子的构效关系以及未来的分子结构设计提出了明确的方向.除了化学键结构与类型对器件性能的影响外, 分子的空间构象也对材料特性及器件性能产生巨大影响. 因此, 近年来的分子设计方面也从分子的空间构型角度进行了较多的研究工作, 形成了较为系统的分子结构拓扑工程(Molecular Topology Engineering, MTE). 具体来说, 分子结构拓扑工程是一种通过精准设计分子空间结构及其连接方式, 构建出分别具有一维线性、二维平面以及空间三维扩展的新型共轭分子, 进而调控分子间相互作用、电子传输路径和材料性能. 如图2所示, 一维分子是指其共轭骨架主要沿着一个空间方向延伸的一类共轭分子, 由若干个芳香亚单元通过C—C单键相连形成, π电子沿线性方向传输[27]; 二维分子则是指其共轭骨架在一个二维平面内扩展和离域的一类共轭分子. 其平坦、刚性的结构容易使二维平面分子之间存在强烈的面对面π-π堆积作用, 形成高度有序的结晶性薄膜, 有利于分子间的电荷传输[28]. 现实中, 由于分子内大基团间的空间位阻, 往往使分子骨架弯曲形成一定的夹角, 完全平面化的二维NFAs几乎没有. 因此在本文中分子内夹角不超过30º的NFAs, 均近似认为是二维平面结构. 类似地, 三维分子是指其共轭骨架通过立体化学设计向三维空间扩展的共轭分子, 这类分子的核心特征是空间维度的突破. 通过引入空间位阻迫使分子结构发生巨大扭曲, 其π电子离域和电荷传输路径不再是线性的或面性的, 而是倾向于形成各向同性的三维网络[29]. 基于共轭分子空间结构成为新型NFAs开发的新发展趋势, 本综述从共轭分子空间结构拓扑扩展的角度出发, 总结了具有一维线性、二维平面型和三维空间型分子的结构特征、分子设计思路、研究进展及其相应的结构-性能关系, 并对后续的分子设计创新进行了展望.
1 一维线性非富勒烯受体分子的研究进展
1.1 线性稠合小分子受体: 从“S”型骨架到“C”型骨架的演变
到目前为止, 一维线性共轭NFAs分子可分为三类: 稠合小分子受体、非稠合小分子受体和寡聚巨分子受体. 线性稠合小分子受体是当前高性能有机太阳能电池的研究主体与效率突破的基石. 其分子拓扑结构的核心特征在于由一个刚性的、平面的梯形稠环结构作为电子给体D核, 两端连接强电子受体A核作为封端, 形成A-D-A的线性构型, 是小分子给体发展的延续.
早期的代表性设计基于C2h对称性的“S”型拓扑骨架, 该类骨架通常由给电子能力的稠环单元构成核心, 其结构特征在于分子主轴呈舒缓的“S”形弯曲(图3左). 这种拓扑结构初步解决了早期受体刚性过强、过度聚集的问题. 如上文所述, 占肖卫课题组[18]开发的ITIC分子是具有里程碑意义的突破, 其分子拓扑核心是一个IDTT单元, 该稠环核具有相对刚性的“S”型骨架, 增强了分子平面性和共轭长度, 拓宽了分子的吸收, 保证了分子的电荷传输. ITIC的成功确立了以线性A-D-A稠合结构为核心的高效NFAs分子思路, 引发了后续如IT-4F[30]、ITIC-Th[31]等衍生物的研究. “S”型骨架的另一典型代表是苯并二噻吩(BDT)类NFAs. BDT核相比于IDT/IDTT核拥有更强的给电子特性, 更好的平面性, 更窄的带隙以及更优越的电荷传输性能. BDT独特的“二维对称性”可以实现分子更精细地调控. 例如在苯环的5位引入烷氧基、烷硫基以及芳基等强给电子侧链, 不仅可以调控溶液加工性能, 而且可以进一步抬高HOMO能级缩窄带隙, 强化吸收特性. 例如车广波组[32]在5位引入噻吩[3,2-b]并噻吩(TT)侧链增强π电子的离域范围, 使BTT- FIC的带隙降至1.47 eV. 同时, BDT核心的高平面结构与TT侧链的垂直堆叠特性提升了分子间的π-π堆叠效率, 使器件的载流子迁移率达1.87× 10-3 cm2•V-1•s-1, 解决了IDT/IDTT核类NFAs带隙宽, 迁移率低的问题. 除了5位, 噻吩环上的2,6位和3,7位也可以进行灵活的原子取代, 实现更精细的带隙调控. 例如郑庆东课题组[34-35]通过在2,6位和3,7位引入噻吩[3,2-b]并吡咯(TP)基团, 形成无sp3杂化碳的M系列受体, 利用吡咯氮原子的强给电子效应, 进一步将带隙压缩至1.30~1.40 eV 区间. 通过利用“邻位侧链”的位阻效应来抑制受体分子的过度自聚, 实现了更紧致的π-π堆积, 从而有效解决了IDTT核系列NFAs堆积较弱, 电荷复合率高的问题, 代表分子如MC7F3[33]和M34[34]等.
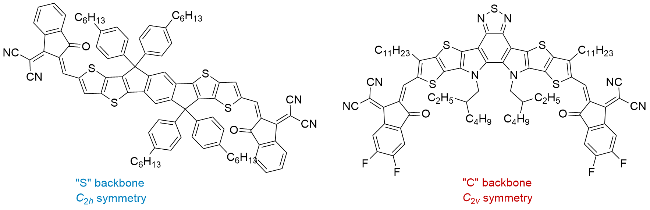
Y系列NFAs的出现打破了“S”型分子骨架设计的路径依赖. 相比于“S”型骨架, 以Y系列NFAs为代表的“C”型分子骨架则表现出更优异的光伏性能, 也是目前主流高效NFAs所采用的分子骨架(图3右). 这种分子骨架使分子呈急剧的“香蕉”形弯曲. 2024~2025年, 郑彦如[35]、黄飞[36]和李永舫[37]团队分别在其研究中揭示了“C”型骨架相对于“S”型、“M”型以及“?”型等其他类型骨架的构象优势, 包括薄膜态下诱导分子形成“J”聚集拓宽分子吸收, 实现更窄的光学带隙; 单晶体积小可被烷基链均匀填充, 自聚集程度弱; 与给体兼容性良好, 能形成紧密的π-π堆积以提升载流子迁移率; 低分子重组能可抑制非辐射衰减等. 值得注意的是, 缺电子A'核对拓宽分子吸收的影响有限, 主要增强分子的聚集行为. 基于“C”型分子骨架, 研究人员通过核心工程和侧链工程进一步提升NFAs的光电及光伏性能, 例如在A'核中引入刚性平面单元强化面朝上(face-on)的分子堆积取 向[38]; 改变侧链的支化位点优化相分离形貌等[39], 这些工作都为高效“C”型稠环骨架的分子设计提供了大量的参考依据.

1.2 线性非稠合小分子受体的研究进展
如图5所示, 非稠合小分子受体是由若干个噻吩/苯或简单并环结构通过C—C偶联形成的一类线性共轭分子, 可看作是将稠环受体分子中的部分并环结构通过裁剪获得. 非稠合小分子受体的优点在于结构简单、多样、柔性且易于合成. 该类受体的首次报道可追溯至 2018 年, 由陈红征团队[40-42]开发的PCIC系列. 之后, 黄辉[43]、薄志山[44]、陈永胜[45]以及侯剑辉[46]团队在此基础上深入研究, 开发出多样的非稠合小分子NFAs材料. 目前基于该类受体的单结光伏器件效率已突破19%[47], 有望超越稠合小分子受体. 针对非稠合小分子受体面临构象易扭转导致的堆积无序, 电压损失高, 迁移率低等问题[40-47], 研究人员提出引入“构象锁”或大位阻侧链以抑制C—C单键扭转, 提升分子的刚性, 进而调控分子的堆积行为, 缓解复合损失.

1.2.1 含“构象锁”的线性非稠合小分子受体光伏性能调控研究
图6展示了含构象锁的非稠合小分子NFAs的化学结构. 2012年, 黄辉与Marks和Facchetti团队[48]在研究场效应晶体管材料时, 提出半导体分子内的某些原子间非共价相互作用, 可以降低芳香亚单元间的夹角, 形成和稠环分子相同的刚性分子, 称之为非共价构象锁(Non Conformational Locks, NoCLs). 之后的数年里, 研究学者通过对NoCLs的深入研究, 证实其可以通过增强分子的刚性, 从而改善器件的电荷传输性能. 郑超团队[49]通过理论计算总结了NoCLs在不同芳环体系的作用规律, 提出了软共轭分子(Soft Conjugated Molecules, SCMs)的概念, 即在固态锁定刚性共平面结构以增强光电性能, 在溶液中因非平面结构提升加工性. 在NoCLs的分类中, 强作用对包括S…N、N…H、S…O和F…S等相互作用, 弱作用对包括F…N、F…H和S…H等相互作用. 强作用对使分子能级深陷, 促进分子内电荷转移(Intramolecular charge transfer, ICT), 引起吸收的显著红移, 与侧链工程、端基工程的协同作用有助于改善分子的堆积行为, 并提高载流子迁移率.. 例如2019年, 薄志山课题[50]开发了含S…O构象锁的DO系列非稠合小分子NFAs. 与不含构象锁的分子DC6-IC相比, 理论计算表明, 构象锁的存在使分子内各亚单元排列在同一平面内, 分子内的二面角几乎为零. 共平面构想增强了分子间的π-π堆积, 使分子的吸收红移了近200 nm. 当选用2-乙基己基作为侧链, 2F-IC作为端基时, DOC2C6-2F与给体构成的共混膜表现出最小的相分离尺寸与均匀有序的堆积形貌, 器件的载流子迁移率达到数量级为10-3 cm•V-1•s-1的高水平. 采用直链或无卤端基的NFAs, 则表现出过强的相分离和较无序的分子堆积, 一定程度上抑制了电荷的分离与传输过程. 由此可以看出, 构象锁还需协同其他分子工程才能系统优化分子的性能. 结合侧链工程, 黄辉课题组[51]报道的NoCA-5和Langa组[52]报道的TCOR2, 实现了分子溶解性与结晶性的良好平衡, 显著提高器件的电荷分离与传输性能, 是目前基于NoCLs调控的最高效的非稠合小分子NFAs.
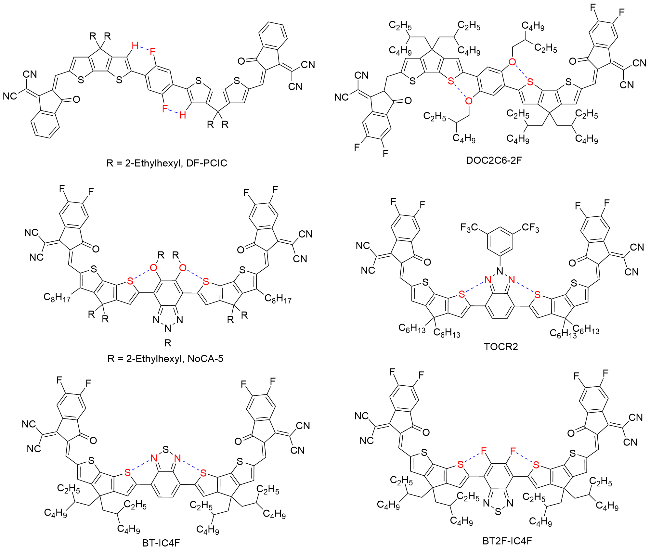
值得关注的是, 虽然在同一芳环体系中可能存在不同作用强度的NoCLs, 但它们形成的优先顺序循序并不遵循“先强后弱”的规律, 更多是从理论计算的角度考虑它们对分子整体自由能的降低效果. 例如理论计算结果显示, 在非稠合小分子受体DF-PCIC中, 优先形成F…H弱作用对而非F…S强作用对; BT系列受体中BT- IC4F分子内仅形成噻吩与苯并噻二唑之间的S…N作用对, 而当氟原子引入后, BT2F-IC4F内却优先形成相对较弱的F…S作用对, 使分子的HOMO能级降低了近0.1 eV[53], 吸收蓝移. 不仅如此, F…S作用对的引入还使分子的结晶性增强, 扩大了活性层的相分离尺寸, 一定程度上电荷分离受到抑制, 导致器件的电流偏低. 研究表明, 尽管构象锁被视为一种有效的结构调控策略, 然而其效果受共轭骨架、侧链、溶剂环境等多因素影响, 构效关系难以通过简单规则预测, 需结合理论计算和原位表征揭示其的动态作用机制, 并反复试验验证. 除此之外, 大多数NoCLs在高温或外界应力下易断裂, 导致构象失稳, 不适用于结构更简单、更柔性的分子. 截至目前, 常用于非稠合小分子NFAs的NoCLs仅包括S…N、S…O、F…H、F…S相互作用. 未来还应尝试开发含F…N、N…H和S…H等相互作用的受体分子, 并总结出这些NoCLs在同一分子骨架中是否存在着相互制约或相互协同的关系, 以便更精准地调控分子性能.
1.2.2 含大位阻侧链的线性非稠合小分子受体光伏性能调控研究
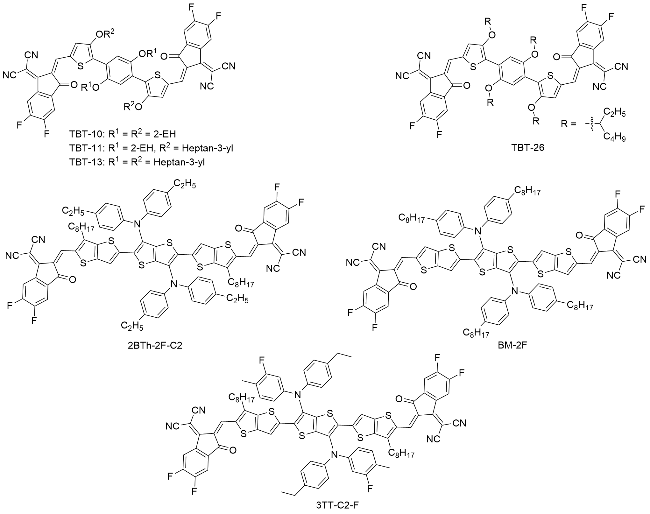
对于噻吩-苯类和寡聚噻吩类等结构更简单的完全非稠合小分子受体, 引入大位阻侧链可替代NoCLs提升分子的刚性. 其中含支链的烷基侧链适用噻吩-苯类受体, 而间位取代的苯基和二苯胺类侧链更适用于寡聚噻吩类受体. 这种侧链选择策略的差异归因于噻吩和苯环本身的空间位阻不同. 具体而言, 噻吩的空间位阻比苯环稍小, 采用位阻较小的烷基类侧链难以缓解C—C单键旋转, 而苯环本身的大位阻容易与苯基侧链发生扭转使侧链垂直于共轭主链之外, 对噻吩几乎不产生位阻. 图7展示了含大位阻侧链非稠合小分子受体的化学结构. 在噻吩-苯类受体中, 支链侧链的支化位点内移可显著增强分子的共平面性, 这在侯剑辉团队[46,54]开发的噻吩-苯-噻吩(TBT)系列受体中有详细的论述. 需要指出的是, 除了大位阻支化侧链, TBT系列分子中的S…O构象锁也对分子的平面性起到了一定的协同作用. 寡聚噻吩类受体中, 薄志山团队揭示了二苯胺侧链相对于间位取代的苯基侧链的优越性, 主要与共轭主链互相垂直的堆叠特性有助于形成三维互穿网络结构, 促进分子间的电荷传输. 代表受体分子包括2BTh-2F-C2[47]、BM-2F[55]、3TT-C2-F[56]等. 其中基于D18:2BTh-2F-C2的器件在界面优化后获得19.02%的转换效率, 是基于线性共轭小分子受体器件的最高效率记录[47].
1.3 寡聚线性巨分子受体的研究进展
除了小分子受体, 寡聚线性巨分子受体近2~3年来也愈发受到关注. 巨分子受体(Giant Molecular Acceptors, GMAs)是由两个或多个小分子受体的亚单元, 通过共轭或非共轭方式连接单元构成的大分子结构. 其优点在于解决了小分子受体扩散系数较高, 形貌不稳定性的问题[57-58]. 在李永舫团队的论述中, 巨分子受体兼具高效小分子受体的宽吸收、适配的能级、优异的成膜性和构象稳定性, 同时避免了聚合物小分子受体因分子量波动存在的批次间重复性差等问题[58-59]. 如图8所示, 为保证π电子可在多个亚基团之间沿线性传输, 本综述讨论的寡聚线性巨分子仅针对“尾对尾”型共轭骨架. 按照π-桥联单元的类型可分为烯/炔基型、芳香基型和烯/炔-芳香杂化型.
如图9所示, 烯/炔基是构建GMAs最简单的一类结构化π-桥联单元, 其与小分子受体亚单元端基上的内侧碳(靠近羰基一侧)相连, 更有利于光伏性能的提升. 这归因于内侧碳相连增强了分子内的共轭效应, 而外侧碳(靠近丙二腈基一侧)相则阻断了π电子在小分子受体亚基团间的传输, 不利于Jsc的提升[60-61]. 从共轭分子共平面性的角度出发, 烯/炔-芳香杂化型π-桥联单元对巨分子的共平面性贡献度最高, 这是由于烯/炔基本身的刚性以及其与芳香基团的共轭效应所致[62]. 烯/炔基型的共平面性则次之, 而芳香型基团本身的位阻易与小分子亚基团形成较大扭角, 分子构象最容易弯曲. 分子的共平面性程度越高, 意味着材料的结晶性越强, 玻璃转化态温度越高, 分子的扩散系数越低, 对器件的热稳定性越有利[63]. 与非稠合小分子受体的调控策略类似, 一些研究表明: 在芳香基型π-桥联单元与小分子受体亚基团间引入分子内NoCLs, 也能提升巨分子的结晶性, 从而促进分子间紧致的π-π堆积, 提升电荷传输性能. 例如Wang[63]与黄辉[64]等分别开发出含Se…F和S…F非共价作用的巨分子受体DIBP3F-Se和DYF-TF, 使器件的载流子迁移率相比于其同类参照物(分别为DIBP3F-S和DY-T)提升超过一倍. 然而无论采用何种连接方式, 一个不容忽视的问题是, 过强的结晶性容易导致分子的过度聚集, 抑制电荷分离. 因此需要利用这一构效关系精准调控寡聚线性巨分子的聚集和扩散行为, 兼顾器件效率与稳定性. 除了π-桥联单元的类型和作用位点, 分子的聚合程度也深刻影响着分子的聚集和扩散行为. 聚合程度越高的巨分子受体凭借其高分子量对低分子扩散系数的增益愈发明显[65], 但同时也存在合成复杂度提升, 可溶液加工性能变差, 分子易过度聚集等问题. 因此就旋涂工艺而言, 较为适宜的寡聚程度应当是三聚或四聚体.
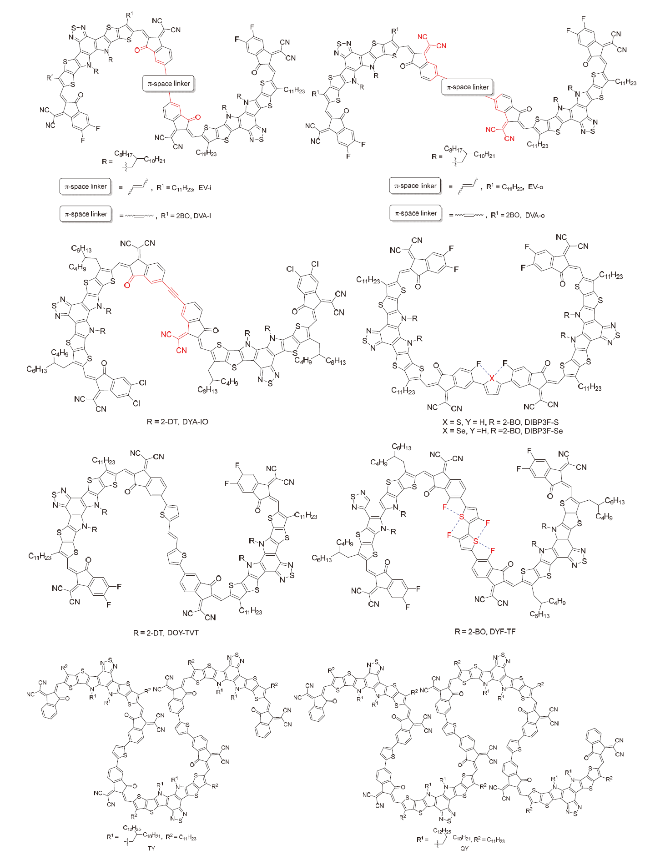
表1展示了上述三类代表性线性NFAs分子的光电及光伏性能. 线性稠合小分子受体引领了OSCs受体材料从传统富勒烯体系向NFAs体系的跨越和从“S”型骨架向“C”型骨架的演变, 将OSC的性能和潜力提升到了一个前所未有的高度. 高效的线性非稠合小分子NFAs结构简单、合成便捷, 兼具高效率与低成本的优势, 但是其固有的小分子尺寸和过于柔性的特性对分子间有序的π-π堆积和电荷传输不利, 需要持续提升分子的刚性. 寡聚巨分子NFAs凭借其低分子扩散系数的优势在构筑高效稳定的活性层方面取得了飞速的进展. 目前大多数基于寡聚线性巨分子受体器件的效率分布在16%~19%之间, 与线性稠合小分子NFAs基本持平, 但明显优于线性非稠合小分子受体. 巨分子NFAs继承了稠合小分子NFAs的刚性骨架, 且大分子量缓解了巨分子在活性层中的扩散趋势, 因此器件效率和稳定性优于所有小分子受体. 鉴于印刷工艺的不断成熟与开发高效大面积光伏组件的需要, 活性层组分对形貌稳定性的要求将不断提升, 分子寡聚化将是未来的发展趋势. 然而巨分子繁琐的合成路线造就了其高昂的成本劣势. 为了实现效率、成本和稳定性的有机统一, 进一步降低稠环小分子NFAs的合成成本, 并开发高效的聚合工艺应当是合成方法学所关注的重点.
表1 线性非富勒烯受体分子的性能参数Table 1 Performance parameters of linear non-fullerene acceptors |
| NFAs | Donor | HOMO/LUMO/eV | Voc/V | Jsc/(mA•cm-2) | FF/% | PCE/% | Ref. |
|---|---|---|---|---|---|---|---|
| BTT-FIC | PM6 | -3.87/-5.64 | 0.953 | 18.54 | 71.6 | 12.65 | [32] |
| MC7F3 | PM1 | -4.06/-5.69 | 0.863 | 25.68 | 79.5 | 17.61 | [33] |
| M34 | PM6 | -3.90/-5.61 | 0.910 | 23.63 | 70.7 | 15.24 | [34] |
| SB16 | PM6 | -4.11/-5.54 | 0.850 | 0.740 | 23.3 | 0.15 | [35] |
| CB16 | PM6 | -3.97/-5.73 | 0.860 | 25.98 | 76.9 | 18.32 | [35] |
| S-F | D18 | -3.87/-5.78 | 0.862 | 23.20 | 77.2 | 15.40 | [36] |
| C-F | D18 | -3.86/-5.78 | 0.921 | 24.00 | 77.1 | 17.00 | [36] |
| S-Cl46-Cl | D18 | -4.01/-5.68 | 0.872 | 10.23 | 34.1 | 3.04 | [37] |
| M-Cl46-Cl | D18 | -3.97/-5.85 | 0.904 | 13.43 | 44.8 | 5.44 | [37] |
| ?-Cl46-Cl | D18 | -3.95/-5.91 | 0.993 | 17.21 | 72.1 | 12.32 | [37] |
| C-Cl46-Cl | D18 | -3.99/-5.79 | 0.922 | 26.50 | 81.6 | 19.94 | [37] |
| DF-PCIC | PBDB-T | -5.49/-3.77 | 0.910 | 15.66 | 72.1 | 10.14 | [40] |
| DOC2C6-2F | PBDB-T | -5.49/-3.83 | 0.850 | 21.35 | 73.2 | 13.24 | [50] |
| NoCA-5 | J52 | -5.43/-3.83 | 0.810 | 26.02 | 69.9 | 14.82 | [51] |
| TCOR2 | P1 | -5.59/-4.02 | 0.870 | 24.22 | 72.0 | 15.17 | [52] |
| BT-IC4F | PBDB-T | -5.89/-4.27 | 0.690 | 21.40 | 66.4 | 9.83 | [53] |
| BT2F-IC4F | PBDB-T | -5.98/-4.31 | 0.670 | 19.43 | 64.7 | 8.45 | [53] |
| TBT-10 | PBQx-TF | -5.68/-3.91 | 0.826 | 12.00 | 46.0 | 4.54 | [54] |
| TBT-11 | PBQx-TF | -5.62/-3.93 | 0.847 | 15.40 | 57.0 | 7.44 | [54] |
| TBT-13 | PBQx-TF | -5.49/-3.97 | 0.798 | 25.90 | 77.9 | 16.10 | [54] |
| TBT-26 | BBQx-TF | -5.54/-4.16 | 0.808 | 26.10 | 80.7 | 17.00 | [46] |
| 2BTh-2F-C2 | D18 | -5.55/-3.98 | 0.910 | 26.71 | 77.9 | 19.02 | [47] |
| BM-2F | J52 | -5.39/-3.99 | 0.810 | 25.25 | 70.7 | 14.53 | [55] |
| 3TT-C2-F | D18 | -5.56/-3.95 | 0.900 | 24.13 | 78.6 | 17.19 | [56] |
| EV-i | PM6 | -5.77/-3.91 | 0.897 | 26.60 | 76.6 | 18.27 | [60] |
| EV-o | PM6 | -5.80/-3.71 | 0.957 | 6.20 | 42.1 | 2.50 | [60] |
| DYA-I | D18 | -5.51/-3.99 | 0.938 | 25.67 | 78.00 | 18.83 | [61] |
| DYA-O | D18 | -5.47/-3.93 | 0.948 | 24.29 | 76.00 | 17.54 | [61] |
| DYA-IO | D18 | -5.49/-3.96 | 0.961 | 23.32 | 73.00 | 16.45 | [61] |
| DIBP3F-S | PM6 | -6.03/-4.29 | 0.901 | 24.86 | 72.00 | 16.11 | [63] |
| DIPB-3F-Se | PM6 | -6.00/-4.27 | 0.917 | 25.92 | 76.10 | 18.09 | [63] |
| DOY-TVT | D18 | -5.80/-3.66 | 0.852 | 27.53 | 77.01 | 18.06 | [62] |
| DYF-TF | D18 | -5.59/-3.63 | 0.939 | 25.82 | 75.3 | 18.26 | [64] |
| DY | PM6 | -5.66/-3.74 | 0.959 | 22.01 | 70.5 | 14.88 | [65] |
| TY | PM6 | -5.64/-3.75 | 0.953 | 23.35 | 73.4 | 16.32 | [65] |
| QY | PM6 | -5.64/-3.76 | 0.937 | 23.20 | 71.2 | 15.47 | [65] |
2 二维平面非富勒烯受体的研究进展
二维平面非富勒烯受体基于平面共轭骨架设计, 扩大了π电子的离域范围. 平面分子的刚性骨架有利于形成连续的电子传输通道, 压缩吸收带隙, 拓宽吸收光谱, 提升消光系数. 通过平面骨架修饰(如卤素取代、杂原子掺杂)可灵活调控分子的能级与带隙. 然而刚性结构容易导致分子过度聚集形成微米级的大相分离畴区, 不利于界面处的电荷分离; 除了PDI外, 常见的二维基团包括萘、芘以及蒽等衍生物. 然而在实际分子骨架的形成过程中, 基团间的空间位阻容易导致构象弯曲形成严格意义上的三维空间结构. 因此本综述讨论的受体分子若其二面角不超过30°, 均可近似认为是二维平面结构.
2.1 基于PDI系列二维受体分子的研究进展
PDI作为典型的二维平面单元在NFAs材料的早期探索及机理研究中起到了试验田的作用. 然而这种单一结构存在着吸收过窄, 堆积过强的缺陷. 研究发现, 基于PDI受体的器件的效率与填充因子(Fill Factor, FF)呈近似正线性相关, 而FF主要由给受体的相容性及相分离决定, 且形态受分子几何结构显著影响. 幸运的是, PDI的湾位活性为其改性修饰提供了一定空间. 随着A-D-A型和A-DA'D-A型NFAs的开发应用, 研究人员尝试将PDI基团作为A核融入到梯形稠环结构中以拓宽吸收并缓解过度聚集的问题(图10). 例如, 钟洪亮 组[66]将氟代PDI作为封端基团与IDTT核稠合形成了A-D-A型二维拓扑受体F-2PDI和F-2PDI-4F. 湾位氟原子的引入使分子因F…F排斥作用呈现出二面角约12º的柔性弯曲, 一定程度上抑制了PDI基团间的过度聚集, 促进了电荷的有效分离. 将PDI作为核心与IDT核稠合可分别形成“S”型和“C”型骨架的A-DA'D-A型NFAs. 有趣的是, “S”型骨架受体anti-PDFC表现出优于“C”型骨架受体syn-PDFC的光伏性能, 二者器件的Jsc和FF参数体现出明显差距[67-68](表2), 这说明在“S”型骨架中侧链的空间位阻更能有效抑制PDI核的过度聚集. 此外, 侧链的空间取向对分子的堆积行为也有显著影响. 研究表明正交的烷基侧链有利于分子间的有序堆积, 而非正交的苯基侧链则导致PDFC-Ph堆积无序, 造成了载流子迁移率过低. 虽然利用稠合策略结合侧链工程可以有效缓解PDI核吸收窄、过度聚集的不足, 然而PDI本身作为大体积基团容易因空间位阻产生分子内构象弯曲, 进而影响到分子的吸收、能级、堆积等一系列光电特性, 使分子调控更加复杂化, 这也限制了它们的进一步应用.
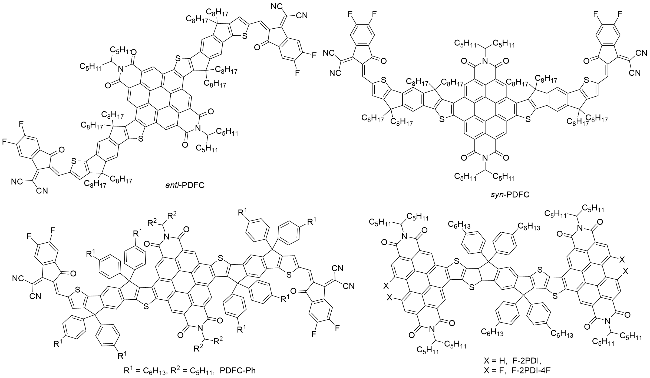
表2 平面非富勒烯受体分子的性能参数Table 2 Performance parameters of planar NFAs |
| NFAs | Donor | HOMO/LUMO/eV | Voc/V | Jsc/(mA•cm-2) | FF/% | PCE/% | Ref. |
|---|---|---|---|---|---|---|---|
| F-2PDI | PBDB-T | -5.52/-3.76 | 1.030 | 11.23 | 60.4 | 7.07 | [66] |
| F-2PDI-4F | PBDB-T | -5.55/-3.81 | 1.000 | 13.42 | 66.8 | 9.05 | [66] |
| anti-PDFC | PM6 | -5.57/-3.85 | 0.970 | 15.93 | 81.3 | 12.56 | [67] |
| anti-PDFC | BTR-Cl | -5.57/-3.84 | 0.972 | 12.09 | 60.2 | 7.08 | [68] |
| syn-PDFC | BTR-Cl | -5.58/-3.83 | 1.008 | 11.09 | 55.4 | 6.19 | [68] |
| PDFC-Ph | BTR-Cl | -5.61/-3.93 | 1.034 | 2.43 | 31.8 | 0.798 | [68] |
| CH8 | PM6 | -5.64/-3.75 | 0.889 | 19.70 | 53.5 | 9.37 | [69] |
| 4A-DFIC | D18-Cl | -5.72/-4.03 | 0.905 | 22.47 | 77.4 | 15.76 | [70] |
| DP-BTP | D18 | -5.67/-3.92 | 0.960 | 22.73 | 69.1 | 15.08 | [71] |
| QD-1 | PM6 | -5.73/-3.80 | 0.895 | 27.52 | 79.0 | 19.46 | [75] |
| DT-6IC | D18 | -5.65/-3.90 | 0.961 | 22.31 | 73.5 | 15.66 | [74] |
| CH26 | PM6 | -3.59/-5.28 | 0.920 | 22.98 | 72.7 | 15.41 | [73] |
2.2 基于Y系列受体二维扩展的研究进展
2.2.1 二维四臂“蝴蝶翼”型NFAs的研究进展
具有D4h对称性的四臂“蝴蝶翼”构型是Y系列受体最常见的二维扩展. 这种结构是以吩嗪为对称中心沿平面扩展的四臂π共轭体系, 电荷可沿着平面内的四个共轭臂方向传输, 具有低分子重组能, 高电荷迁移率的优点. 由于Y系列受体兼具的内链和外链, 这类二维受体的结晶性与溶解性更易通过增加或减少链长进行调控. 目前已报道的二维平面核心包括苯核、萘核和芘核(图11). 其中以萘为平面核心构筑的二维NFAs取得了最优的光伏性能, 成为二维平面NFAs里的最高记录. 2022年, 陈永胜[69]和丁黎明课题组[70]分别开发出受体分子CH8和4A-DFIC. 其中D18-Cl:4A-DFIC的活性层较PM6:CH8表现出更紧致的堆积行为, 使器件获得了更优的光伏性能. 而CH8因侧链过长造成分子的堆积较弱, 器件的整体性能偏低. 同年, 李韦伟组[71]在核心中引入芘核合成了受体DP-BTP. 芘核的引入增强了分子的结晶性, 并促进了分子间的有序堆积. 作者利用该分子的高结晶性调控基于D18:N3的活性层形貌, 并获得了基于D18:N3:DP-BTP三元器件19.07%的初始转换效率. DP-BTP的高结晶性还可锚定N3, 并抑制其在薄膜中的迁移, 提升器件的热稳定性. 使器件在80 ℃持续加热600 h后效率下降仅10%左右. 陈永胜课题组[72]在CH8的基础上引入萘核, 合成了受体QD-1, 高平面性和中心对称结构促进有序分子堆积, 使基于PM6:QD-1的二元器件FF值达到了79.01%的高水平. 将QD-1作为第三组分掺杂到PM6:BTP-eC9体系中, 发现QD-1还可抑制BTP-eC9的过度聚集, 减少电荷复合, 最终获得了基于20.19%三元器件效率. 从目前已有的报道来看, 四臂二维NFAs的空间构型仍有较大的优化空间. 对于CH8、4A-DFIC和DP-BTP三个分子需要进一步的侧链工程平衡分子的溶解性与结晶性; 除此之外, 可进一步开发诸如蒽核、芴核、苯并二噻吩核等二维核心, 对分子的自组装行为进一步调控.
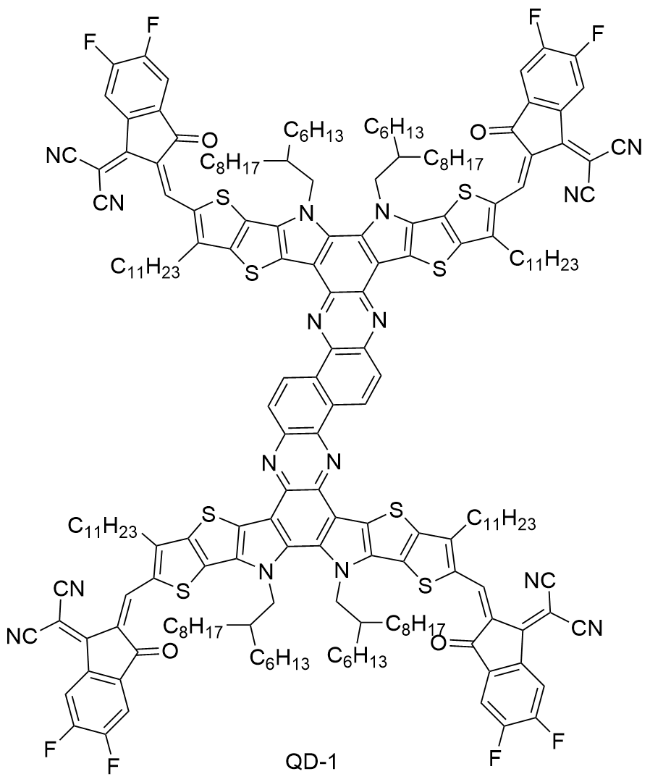
2.2.2 准平面六臂“螺旋桨”型NFAs的研究进展
除了四臂“蝴蝶翼”构型, D3h对称性的准平面六臂“螺旋桨”构型也展现出一定的应用潜力(图12). 这种以苯环、三亚苯为核心, 聚合三个Y系列受体亚单元构成的准平面巨分子具有更高的摩尔消光系数和高玻璃转化态温度. 通过极端二维共轭扩展可显著抬高分子的能级, 有助于构建高开路电压(Voc)器件. 需要指出的是, 六臂拓扑增加了核心与亚单元以及亚单元之间的空间位阻, 从而在热应力下极易造成构型扭曲并破坏平面结构. 虽然这样可以一定程度上提升分子的溶解度, 但不利于分子间的紧密堆积. 例如陈永胜课题组[73]开发的六臂准平面受体CH26获得了0.92 V的高Voc器件, 然而低Jsc和低FF却限制了性能的进一步提升, 器件仅获得 15.41%的转换效率. 类似的结果在丁黎明和李韦伟组[74]报道的六臂受体DT-6IC中也有所体现. 研究发现DT-6IC分子这种“三叶片螺旋桨”的扭曲结构导致薄膜结晶性与π-π相处作用弱于Y系列受体N3, 因而造成了过多的双分子复合, 拉低了器件的Jsc和FF. 当DT-6IC作为第三组分添加到D18:N3体系中时, 却能延缓N3的结晶速度, 并形成均匀的互穿网络, 从而降低相分离尺寸并保证了有效的电荷分离.
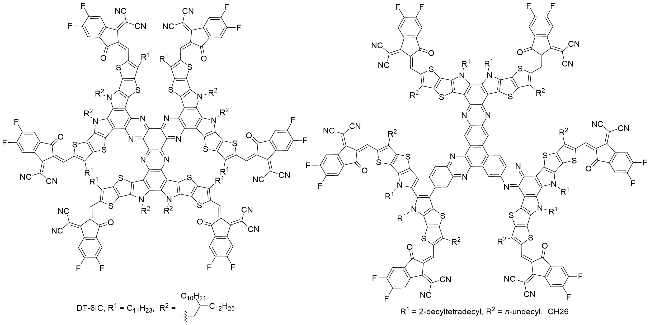
表2列出了相关二维NFAs的光电和光伏性能数据. 相比于一维和三维受体分子, 二维受体的关注度相对较少. 一方面, 如何通过结构设计防止二维结构弯曲形成三维骨架, 缺乏明确的理论指导; 另一方面, 开发多样化的二维核心更需要合成方法学的突破. 由表2可以看出, 尽管通过稠合拓展与侧链工程缓解了PDI吸收窄、易聚集的缺陷, PDI类二维NFAs的光伏性能仍明显低于Y系列二维NFAs. 本文认为四臂“蝴蝶翼”骨架将成为Y系列NFAs主流的二维拓扑结构. 其优点在于分子平面性强不易发生弯曲; 高度对称性的平面易形成面朝上的分子取向和紧致的π-π堆积; 溶解性和结晶性之间的平衡也可通过灵活的侧链工程进行精细调控, 同时保留Y系列NFAs能级和带隙可调的优点. “蝴蝶翼”骨架作为高效NFAs (PCE>19%)的案例已有报道. 六臂“螺旋桨”结构易发生弯曲破坏分子的平面性, 当前可能不适用于高效二元体系的构建. 相反地, 其多臂共轭结构带来的能级提升更有助于构建高Voc器件或改善原有二元体系的相分离结构, 因而可能更适合作为第三组分添加到原有二元体系中.
3 三维空间非富勒烯受体分子的研究进展
三维非富勒烯受体的形成基于大位阻共轭基团之间相互作用引起的结构弯曲. 这类受体通过构建立体共轭网络, 突破了一维线性共轭和二维平面共轭的结构局限. 立体位阻和多维度堆积调控可实现“适度堆积”与“良好溶解”的平衡, 保留分子能级和带隙可调, 以及柔性等优点, 但也会因分子骨架的过度弯曲引起分子的无序堆积, 造成大量的电荷复合. 本综述所讨论的三维受体包括螺环受体分子以及巨分子受体.
3.1 三维螺环受体分子的研究进展
作为早期替代ITIC类受体的初步尝试, 螺环共轭骨架是将两个刚性芳香砌块通过sp3碳原子相连形成的正交结构(图13). 相比于ITIC衍生物及线性受体分子, 螺环小分子受体实现了π共轭体系的三维空间拓展, 为分子内的电荷传输效应提供了多维传输通道, 同时由于两个刚性平面之间的大位阻也可抑制分子间的过度聚集, 给予了螺环小分子受体一定的可溶液加工性能. 在OSCs活性层中, 形貌问题是限制该类受体光伏性能提升的主要因素, 这归因于其过于扭曲的构象造成分子间的弱堆积性能和界面处的电荷复合. 例如陈永胜团 队[75]开发的四臂A-π-D-π-A型受体SFTTIC的摩尔消光系数高达3.12×105 L•mol-1•cm-1, 但当以PBDB-T和PTB7-Th分别作为给体材料时, 器件效率仅为5.66%和4.65%. 从低Jsc和低FF可以看出, 共混摸的相分离尺寸过小, 给受体分子间的π-π堆积较弱, 载流子迁移率低. 研究发现, 当对活性层进行热退火处理或使用强结晶性给体材料时, 可明显提升器件的载流子迁移率, 因此相分离尺寸相对较大的PBDB-T:SFTTIC体系获得了较高的Jsc和FF. 类似的结果在林昳课题组[76]开发的SCT系列A-π-D-π-A型受体中也有体现. 在研究中, 本课题组团队发现所有SCT系列受体分子的薄膜吸收峰较CPDT系列线性受体分子蓝移了25~50 nm, 进一步证实, 相比于线性分子, 螺环分子间的π-π堆积作用较弱. 其中SCT-(TFIC)4因其相对较窄的光学带隙和较强的分子间堆积, 获得了相对较优的光伏性能, 但器件的Jsc和FF明显低于作为参照的线性受体分子CPDT-(TFIC)2. 为了进一步验证构效关系, 林昳课题组[77]在SCT骨架的基础上, 设计合成了一种含双螺氧杂䓬结构的DSOCT系列受体, 该结构含有一个呈船型结构的氧杂䓬七元环, 位于2,7位的两个环戊并二噻吩(CPDT)单元夹角互为72.31°, 形成了更扭曲的分子构象. 尽管分子的消光系数超过了2.0×105 L•mol-1•cm-1, 我们认为扭曲的分子构象造成了分子间更弱的堆积性能, 进一步限制了Jsc和FF, 使器件整体效率不足2%. 因此, 提升螺环分子的刚性或可一定程度上强化分子间的堆积, 提升电荷传输性能. 例如, 杨阳课题组[78]将刚性的梯形稠环结构引入到SBF核中, 合成了螺环受体SFIC-Cl, 使器件的载流子迁移率达到2.24×10-3 cm2•V-1•s-1, 从而有效提升了Jsc, 并最终获得了10.16%的最高效率记录.
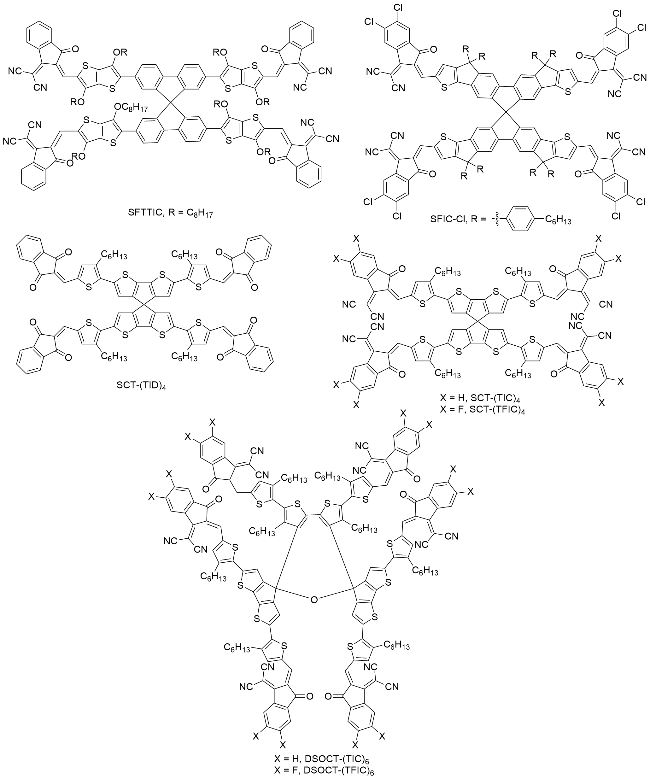
3.2 三维巨分子受体的研究进展
三维巨分子受体是近2~3年受到广泛关注的一类非富勒烯受体, 这类拓扑结构继承了线性巨分子受体玻璃转化态温度高、扩散系数低、形貌稳定性高的优点. 由于核心与亚单元间的弯曲更为明显, 分子构象多呈“星型”或“树枝型”(图14), 有利于抑制分子间的过度聚集, 并形成三维结构的互穿网络堆积形貌, 提升电荷传输性能. 与线性寡聚巨分子和二维分子相同, 高效三维巨分子的骨架是以Y系列NFAs为亚单元的分子设计. 例如李永舫[79]与Kim课题组[80]分别开发的三臂星型巨分子受体GT-s和TYT-S, 均具有较高的玻璃转化态温度(Tg>200 ℃), 在热应力下具有稳定的相分离形貌. 连续光照1460 h和2600 h后, 器件效率仅分别衰减约10%和20%. 在分子内引入非共价相互作用, 也可进一步提升材料的玻璃转化态温度, 优化分子的堆积行为. 包西昌课题组报道了以苯和三嗪为核心, 噻吩为桥的受体3BY和3QY[81]. 研究发现3QY分子内的N…H非共价相互作用提升了三嗪与噻吩的共平面性, 强化了分子间面朝上的堆积取向, 使薄膜表面平整, 纤维状相分离均匀, 提升了器件的电子迁移率, 并实现了19.27%的转换效率. 同时, N…H非共价相互作用提升了分子的结晶性, 使3QY的玻璃转化态温度较3BY提高了近30 ℃, 器件效率保持率T80%>3000 h. 然而非共价作用并非完全能对三维巨对分子NFAs的玻璃转化态温度及堆积行为起到优化作用. 例如刘烽课题组基于三臂星型结构, 将Qx系列受体的核心与苯并三噻吩相连, 并通过在核心和端基分别引入氟原子或氯原子, 合成了三叶草状巨分子受体T-Qx-15F、T-Qx-12Cl3F和T-Qx- 15Cl[82]. 研究表明, 虽然F…H非共价作用会使分子维持平面构象, 但过度平面性导致分子聚集失控, 阻碍电荷传输, 并加剧电荷复合; 同时因缺乏空间位阻屏障致使分子在热应力下沿堆叠方向轻微滑动, 反而降低玻璃转化态温度. 失去F…H作用后, 无构象锁的T-Qx-15Cl因其大扭转角(≈40º)所形成的三维空间位阻, 不仅可缓解分子间的过度聚集, 还可抑制分子间的滑动, 使其不易因热应力发生相变(Tg=188 ℃), 从而维持稳定的电荷传输通道. 这种设计兼顾了高效电荷传输与形态稳定性, 将基于PM6:T-Qx-15Cl的器件效率推至20%, 并在85 ℃退火250 h后效率保持率超98%. 因此可以看出, 是否应当在三维巨分子内引入构象锁, 取决于其能否促进分子间电荷传输的同时, 抑制分子的扩散. 从当前报道来看, 似乎“核心-核心”相连的三叶草结构是三维巨分子受体的最优设计. 需要指出的是, 虽然有GT-s和TYT-S的相继报道, 但尚未有针对性的研究指出“核 心-端基”的连接方式一定劣于“核心-核心”相连, 需要在后续的研究中进行补充讨论.
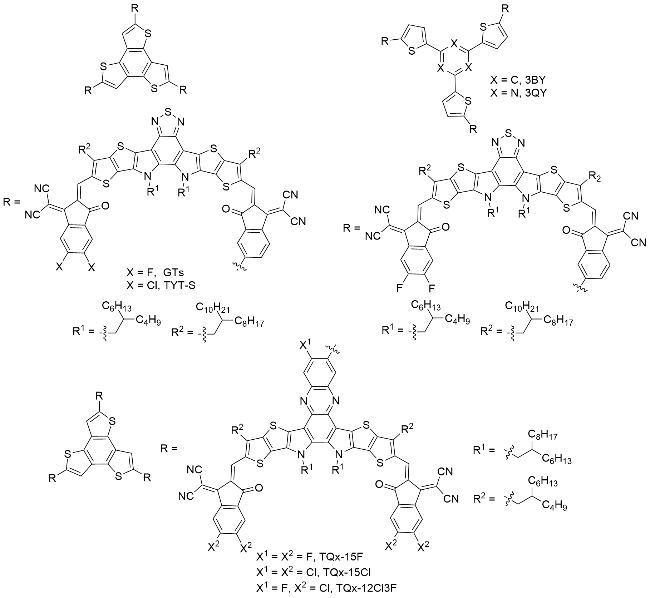
除了核心, 亚基团的数目及空间排布对分子的扩散和堆积行为也产生直接影响. 亚基团数目的增加可以扩大相对分子质量以及分子间空间位阻, 有利于玻璃转化态温度的提升, 抑制分子的扩散行为. 同时也会降低材料的溶解度, 造成分子过度聚集和无序堆积等问题(图15). 例如李韦伟组探究了以苯环为核, 1~6个Y受体亚基团的为臂的树枝受体SPn-Ph系列的光伏性能[83]. 研究发现随着亚基团数的增加, 共混薄膜的相分离尺寸逐渐增大. 其中在基于PM6:SP3-Ph和PM6:SP4-Ph的活性层实现了大小适中的相分离尺寸(8~10 nm)和紧致规则的面朝上π-π堆积, 有效提升了载流子迁移率. 而在基于PM6:SPn-Ph (n=3, 4)的二元器件中, PM6:SP6-Ph体系因相分离尺寸过大(10.99 nm)和分子堆积无序, 导致激子复合加剧. 虽然开路电压有所提升, 但载流子迁移率却大幅下降. 在器件稳定性方面, SP6-Ph因最大的分子质量和最大的分子间空间位阻, 提高了自身的玻璃转化态温度, 抑制了分子的热扩散, 热稳定性最优. 折叠构象作为三维巨分子的独特结构, 可以缩短亚基团间的空间距离, 并形成分子内π-π相互作用, 从而引发电子态耦合效应, 增强对太阳光谱的利用效率. 此外, 折叠构象通过改变分子的拓扑结构, 直接影响分子在薄膜中的聚集与扩散过程, 形成更利于载流子传输的稳定结构. 例如张志国团队[84]报道了两种基于噻吩异构核工程的三维巨分子受体TDY-α和TDY-β. 这两种分子是以异构噻吩二羧酸酯为核, 通过长烷基侧链将两个Y6亚单元共价束缚形成的折叠结构. 研究表明, 两个亚单元互相平行, 强化了分子间面朝上的堆积取向, 有效地平衡了空穴/电子传输(μh/μe≈1); 同时分子内的π-π相互作用, 提升了分子的刚性, 有效限制了分子的热运动, 减少了初始衰减(TDY-α前70 h衰减仅5%, TDY-β为8%, Y6达15%), 最终协同实现器件的高效率与超长稳定性.
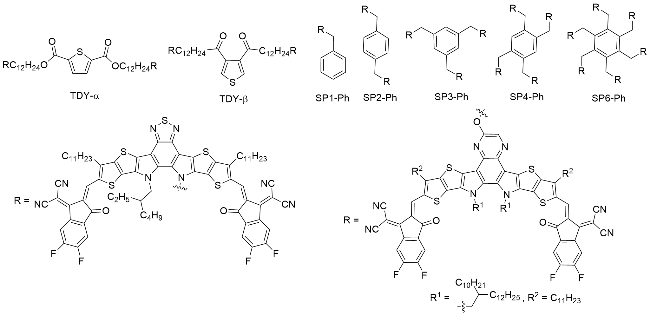
表3列出了相关三维NFAs的光电和光伏性能数据. 由表3可以看出, 以Y系列受体为亚单元的巨分子依然领跑三维NFAs的光伏性能. 螺环小分子因其良好的电荷传输性能, 常被用作传输层材料[85-86]. 当螺环小分子作为活性层的受体材料时, 器件性能普遍不理想. 虽然没有单晶表征揭示螺环受体分子的堆积行为, 但是研究显示螺环受体分子的薄膜吸收较线性分子出现明显蓝移, 且基于螺环受体分子的活性层普遍具有不平衡的空穴/电子迁移率. 因此, 正交扭曲的构象可能不利于分子间的有序堆积, 而提高分子刚性则有望改善这一结构缺陷. 三维巨分子受体通过核心与亚基团之间的空间位阻, 使分子弯曲形成三维空间的互穿网络, 为电荷提供更稳定的传输通道. 其中核的光电特性、亚基团的数量与空间排布均会对受体分子的扩散和堆积行为产生显著影响. 折叠构象通过拉近亚基团间的空间距离, 使分子内外均形成规则紧致的π-π堆积, 增强了分子的刚性. 长烷基侧链的存在则抑制了分子间过度聚集, 在溶解性和结晶性之间保持最优平衡, 既促进了电荷传输性能, 也抑制了分子的热运动, 实现器件效率与稳定性的统一. 多臂构象虽然能通过增加分子间的空间位阻抑制分子的热运动, 从而提升器件的稳定性, 然而亚基团数过多会降低分子的溶解性, 并造成分子过度聚集, 阻碍电荷分离. 研究表明最优的亚基团数目应为3~4个.
表3 三维空间非富勒烯受体分子性能参数Table 3 Performance parameters of 3D spatial non-fullerene acceptors |
| NFAs | Donor | HOMO/LUMO (eV) | Voc/V | Jsc/(mA•cm-2) | FF/% | PCE/% | Ref. |
|---|---|---|---|---|---|---|---|
| SFTTIC | PBDB-T | -5.46/-3.65 | 0.956 | 9.28 | 63.8 | 5.66 | [75] |
| SFIC-Cl | PBDB-T | -5.85/-4.07 | 0.920 | 15.79 | 69.9 | 10.16 | [78] |
| SCT-(TID)4 | PTB7-Th | -5.40/-3.93 | 0.701 | 1.21 | 31.9 | 0.27 | [76] |
| SCT-(TIC)4 | PTB7-Th | -5.45/-4.04 | 0.780 | 6.06 | 69.1 | 2.79 | [76] |
| SCT-(TFIC)4 | PM6 | -5.57/-4.18 | 0.692 | 11.49 | 47.0 | 3.73 | [76] |
| DSOCT-(TIC)6 | PM6 | -5.58/-4.12 | 0.84 | 1.82 | 0.31 | 0.47 | [77] |
| DSOCT-(TFIC)6 | PM6 | -5.67/-4.27 | 0.78 | 4.75 | 0.48 | 1.39 | [77] |
| GTs | PM6 | -5.78/-3.95 | 0.935 | 24.66 | 78.3 | 18.05 | [79] |
| TYT-S | D18 | -5.66/-4.24 | 0.964 | 25.18 | 77.0 | 18.61 | [80] |
| T-Qx-15Cl | PM6 | -5.56/-3.86 | 0.935 | 26.90 | 79.9 | 20.10 | [82] |
| T-Qx-15F | PM6 | -5.54/-3.84 | 0.946 | 24.20 | 75.8 | 17.30 | [82] |
| T-Qx-12Cl3F | PM6 | -5.57/-3.86 | 0.932 | 26.30 | 78.3 | 19.20 | [82] |
| 3BY | PM6 | -5.65/-3.80 | 0.969 | 23.92 | 76.6 | 17.75 | [81] |
| 3QY | PM6 | -5.67/-3.83 | 0.951 | 26.36 | 76.9 | 19.27 | [81] |
| SP1-Ph | PM6 | -5.74/-3.89 | 0.951 | 21.13 | 59.7 | 11.99 | [83] |
| SP2-Ph | PM6 | -5.78/-3.89 | 0.945 | 21.47 | 56.7 | 11.51 | [83] |
| SP3-Ph | PM6 | -5.73/-3.89 | 0.937 | 23.95 | 71.6 | 16.07 | [83] |
| SP4-Ph | PM6 | -5.74/-3.91 | 0.926 | 23.90 | 72.3 | 16.10 | [83] |
| SP6-Ph | PM6 | -5.71/-3.89 | 0.913 | 18.95 | 51.3 | 8.87 | [83] |
| TDY-α | PM6 | -5.69/-3.92 | 0.864 | 26.90 | 78.0 | 18.10 | [84] |
| TDY-β | PM6 | -5.76/-3.96 | 0.849 | 26.10 | 76.6 | 17.00 | [84] |
4 总结与展望
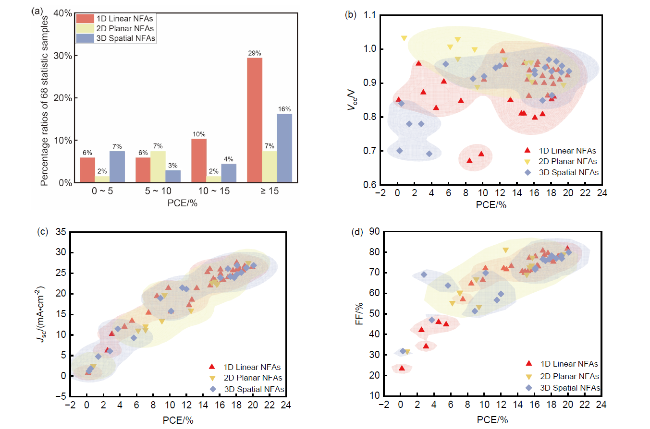
本文系统综述了有机太阳能电池非富勒烯受体分子结构拓扑的研究进展. 从一维线性、二维平面到三维空间结构的设计与优化, 全面探讨了不同空间结构的非富勒烯受体的光电、光伏性能变化规律. 由图16a可以看出, 在68份统计样本中, 分别有29%的一维线性NFAs和16%的三维空间NFAs所制备的光伏器件效率超过了15%. 这主要归因于Y系列线性稠合小分子NFAs的主导地位与分子寡聚化的发展趋势. 线性稠合小分子是当前被研究最广的一类NFAs. 其中“C”型骨架被证实是拓宽分子吸收, 促进分子间有序紧致的π-π堆积, 提升载流子迁移率的关键, 也是线性分子设计中所采用的主流骨架. 然而小分子易扩散的特性极易造成活性层形貌的相变, 器件的稳定性仍不理想. 在出现创新性的分子结构之前, 未来一段时间仍将是以Y系列NFAs的改性修饰为主, 并致力于打破现有的效率记录以及深度的机理研究; 线性非稠合小分子NFAs因较低的合成成本、丰富的结构多样性和良好的光伏性能等优势发展迅速, 然而这类受体分子的普遍柔性更容易引起构象的扭曲和分子间的无序堆积, 导致光学带隙增大和电荷的大量复合. 提高分子的刚性依旧是未来结构优化的主体思路, 需要进一步开发含潜在构象锁(包括未报道的N…H、F…H等相互作用)的非稠合小分子NFAs, 明确多种构象锁分别在同一和不同分子骨架中影响分子刚性的优先顺序, 形成可预测的构效关系; 随着印刷工艺的逐渐成熟与大面积器件需求的增长, 对活性层形貌的稳定性提出了更高的要求, 分子寡聚化成为新的发展趋势. 以Y系列NFAs为亚单元的寡聚线性和三维巨分子NFAs, 因相对可控的分子堆积与扩散行为, 在器件高效率和高稳定性方面均取得了卓越的成效, 有望成为率先商业化的光伏材料. 值得关注的是, 分子多聚化不仅容易造成分子团聚形成大尺寸相分离形貌, 还会因化学选择性变差提高合成成本, 因此三聚体或四聚体或为最优聚合度. 二维平面NFAs继承了线性稠合小分子NFAs的刚性结构, 其共轭链沿平面方向扩展, 提高了消光系数, 同时引导分子间形成取向单一的有序堆积. 基于Y系列NFAs的四臂“蝴蝶翼”和六臂“螺旋桨”骨架, 引领了高效的光伏性能, 使二维NFAs凸显其应用潜力. 为了避免结构弯曲, 适当减少共轭臂数, 增大分子内的夹角或可维持其刚性的平面. 在后续的研究中可关注蒽核、苯并二噻吩核以及吩呐烯核的二维功能化修饰. 螺环类NFAs的光伏性能表现欠佳, 这或是正交结构本身易引起分子构象扭曲和无序堆积导致带隙增大, π-π堆积较弱, 电荷大量复合. 增强分子的刚性虽可一定程度上缓解这些问题, 但由于会导致材料合成成本大幅上升, 其性价比不及非稠合小分子NFAs. 因此, 未来螺环类分子或将继续作为界面传输层材料, 活性层将不再是主流.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
图16 (a)基于多维NFAs器件的效率统计柱状图; (b)多维NFAs的效率-开路电压关系散点统计图; (c)多维NFAs的效率-短路电流密度关系散点统计图; (d)多维NFAs的效率-填充因子关系散点统计图Figure 16 (a) Bar charts of PCEs statistics for multidimensional NFAs based devices; (b) Scatter statistics of PCE-Voc correlations for multidimensional NFAs; (c) Scatter statistics of PCE-Jsc correlations for multidimensional NFAs; (d) Scatter statistics of PCE-FF correlations for multidimensional NFAs |
如图16b~16d所示, 除少数样本外, 基于各类拓扑结构的NFA 器件, 其PCE与Jsc和FF均呈明显正相关关系, 表明无论哪种空间结构, 给受体分子间的能级匹配度, 以及给受体分子间的堆积方式, 实际影响器件电荷的分离与传输过程. 在PCE相近时, 基于三维空间NFAs的器件开路电压略高于一维线性和二维平面NFAs, 而一维线性NFAs的电流表现略胜一筹. 这一现象可归因于: 三维拓扑结构具有更大的空间位阻, 且分子间呈现相对无定形的堆积, 可略微抬升分子的LUMO能级; 而线性分子间形成更规整、有序且致密的π-π堆积,能够为电荷传输提供持续且稳定的通道. 因此, 对于一维线性NFAs, 除了持续提升分子的刚性, 还需通过界面工程和互相掺杂等手段, 提高器件的Voc. 而对于三维巨分子NFAs, 应当考虑核心与亚单元间的区域调控, 以实现分子间更规整、致密的面朝上π-π堆积, 以提高分子间的电荷传输性能. 基于二维平面NFAs的器件, 其Voc与PCE呈较明显的负相关, 表明适当牺牲一定开路电压, 可以换取更大幅度的电流增益, 从而带动器件性能的整体提升. 因此持续提升二维分子的平面性, 并优化其相分离行为; 或开发低能级、窄带隙的新型二维NFAs, 以促进给受体界面处的电荷分离, 是今后的发展方向.
随着合成方法学以及人工智能的不断发展, 我们期待运用更多绿色合成手段, 切实降低NFAs的开发成本, 并基于空间拓扑开发出更多样的结构. 我们同时期待能够基于现有的文献, 总结建立所有NFAs结构及性能大数据库, 进而运用深度学习手段, 指导后续的分子设计, 推动OSCs向市场化迈进.
(Cheng, F.)