Chinese Journal of Organic Chemistry ›› 2026, Vol. 46 ›› Issue (5): 2121-2128.DOI: 10.6023/cjoc202511003 Previous Articles Next Articles
ARTICLES
张帆a, 郭松a, 杨维成a,b,c,*( ), 赵基钢d,*(), 罗勇b,c
), 赵基钢d,*(), 罗勇b,c
收稿日期:2025-12-24
修回日期:2026-01-13
发布日期:2026-02-06
基金资助:
Fan Zhanga, Song Guoa, Weicheng Yanga,b,c,*(), Jigang Zhaod,*(), Yong Luob,c
Received:2025-12-24
Revised:2026-01-13
Published:2026-02-06
Contact:
* E-mail: ywcjc@163.com;
zjg@ecust.edu.cn
Supported by:Share
Fan Zhang, Song Guo, Weicheng Yang, Jigang Zhao, Yong Luo. Analysis of the Nitration Process of Trifluoromethoxybenzene Based on Density Functional Theory (DFT) Calculations[J]. Chinese Journal of Organic Chemistry, 2026, 46(5): 2121-2128.
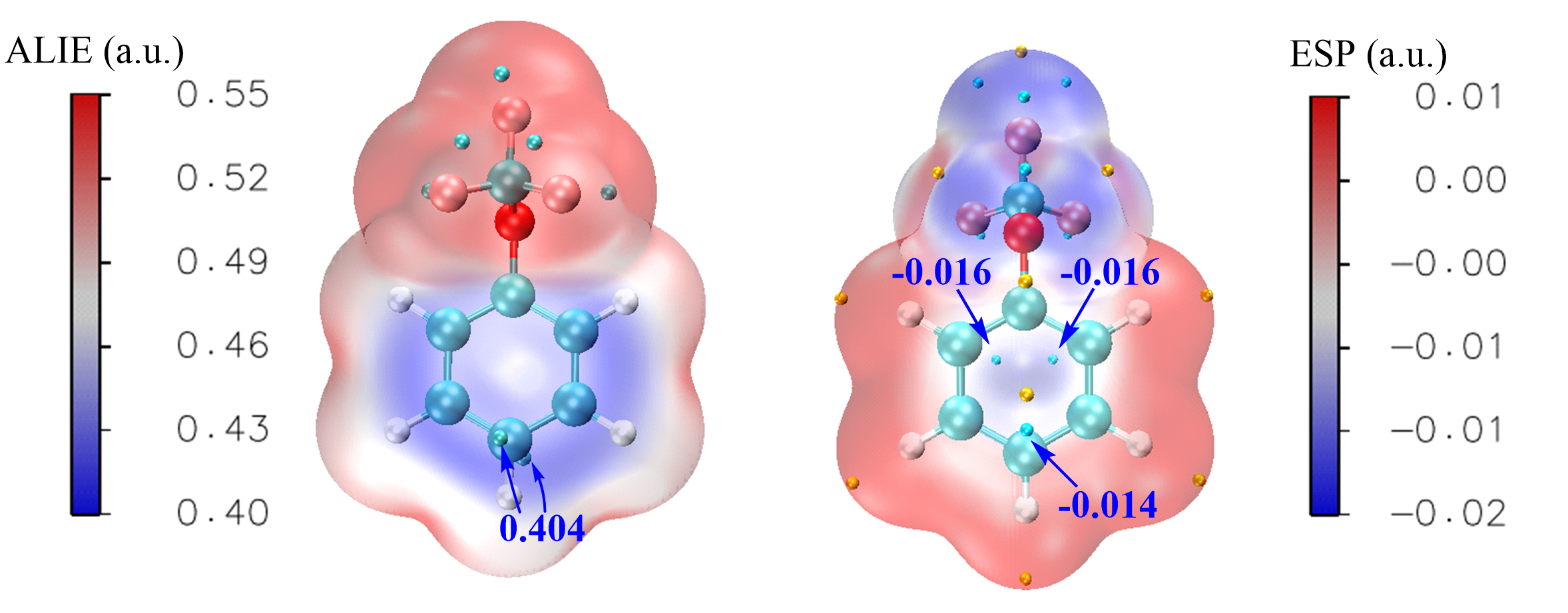
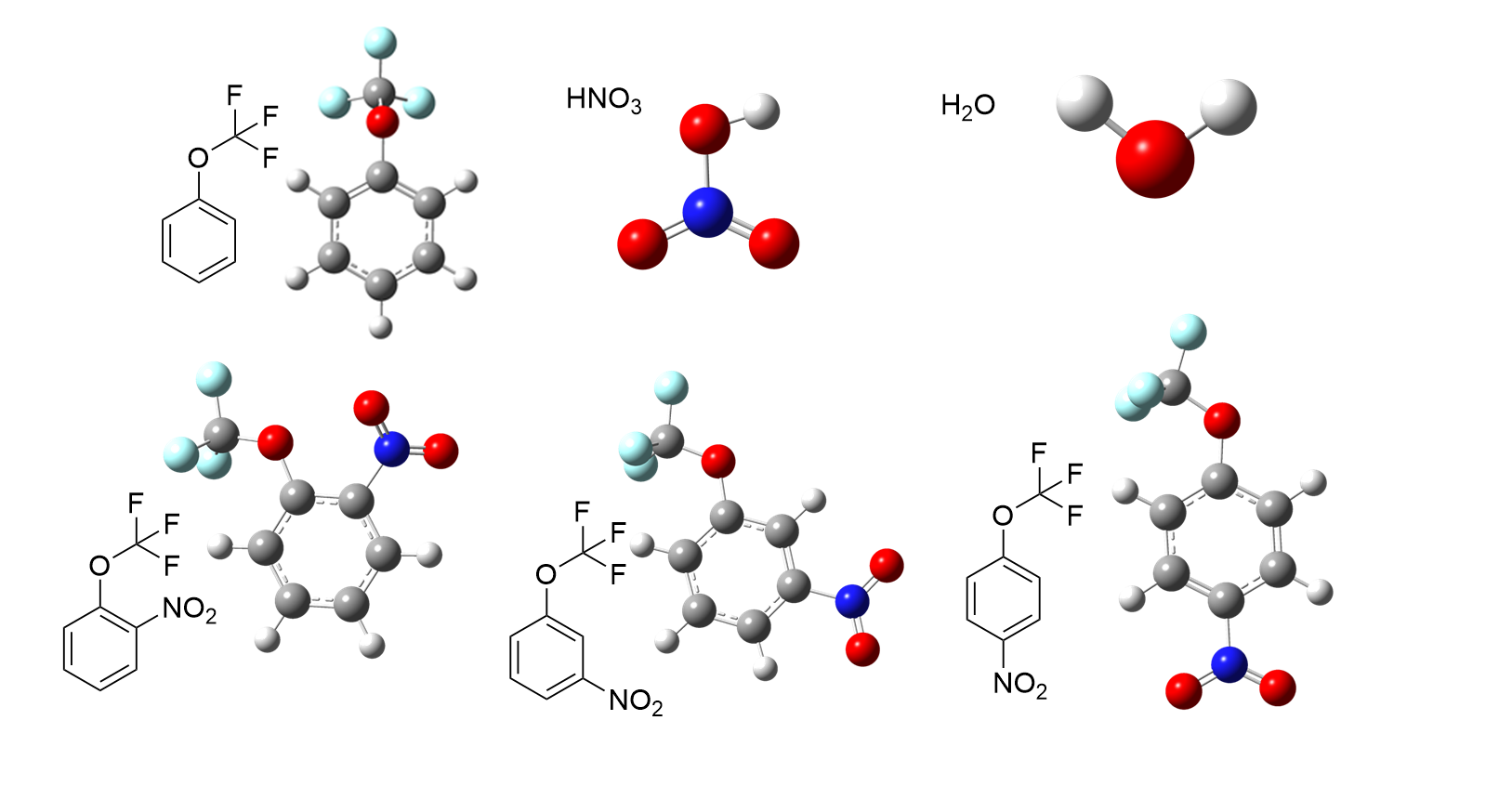
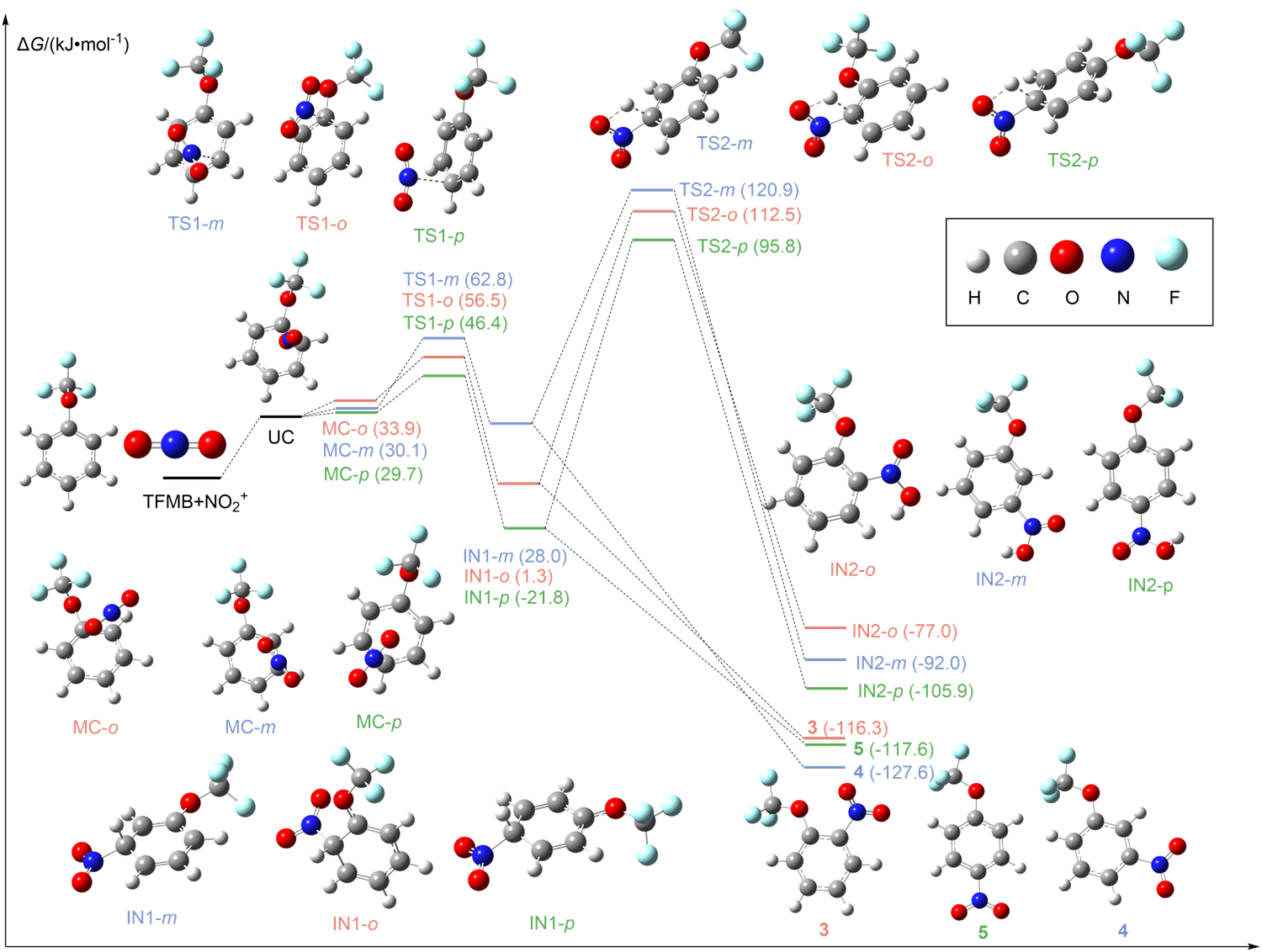
| Compd. | εele/a.u. | ZPE/a.u. | H(0)/a.u. | H(T)/a.u. | S/(J∙mol-1∙K-1) | G(T)/a.u. |
|---|---|---|---|---|---|---|
| TFMB | -644.5139 | 0.1069 | -644.4069 | -644.3973 | 386.137 | -644.4411 |
| HNO3 | -280.8910 | 0.0263 | -280.8647 | -280.8603 | 265.052 | -280.8904 |
| H2O | -76.4320 | 0.0208 | -76.4111 | -76.4073 | 188.644 | -76.4288 |
| 3 | -849.0093 | 0.1100 | -848.8994 | -848.8873 | 436.349 | -848.9368 |
| 4 | -849.0185 | 0.1096 | -848.9089 | -848.8968 | 440.178 | -848.9467 |
| 5 | -849.0206 | 0.1097 | -848.9109 | -848.8988 | 436.040 | -848.9483 |
| Compd. | εele/a.u. | ZPE/a.u. | H(0)/a.u. | H(T)/a.u. | S/(J∙mol-1∙K-1) | G(T)/a.u. |
|---|---|---|---|---|---|---|
| TFMB | -644.5139 | 0.1069 | -644.4069 | -644.3973 | 386.137 | -644.4411 |
| HNO3 | -280.8910 | 0.0263 | -280.8647 | -280.8603 | 265.052 | -280.8904 |
| H2O | -76.4320 | 0.0208 | -76.4111 | -76.4073 | 188.644 | -76.4288 |
| 3 | -849.0093 | 0.1100 | -848.8994 | -848.8873 | 436.349 | -848.9368 |
| 4 | -849.0185 | 0.1096 | -848.9089 | -848.8968 | 440.178 | -848.9467 |
| 5 | -849.0206 | 0.1097 | -848.9109 | -848.8988 | 436.040 | -848.9483 |
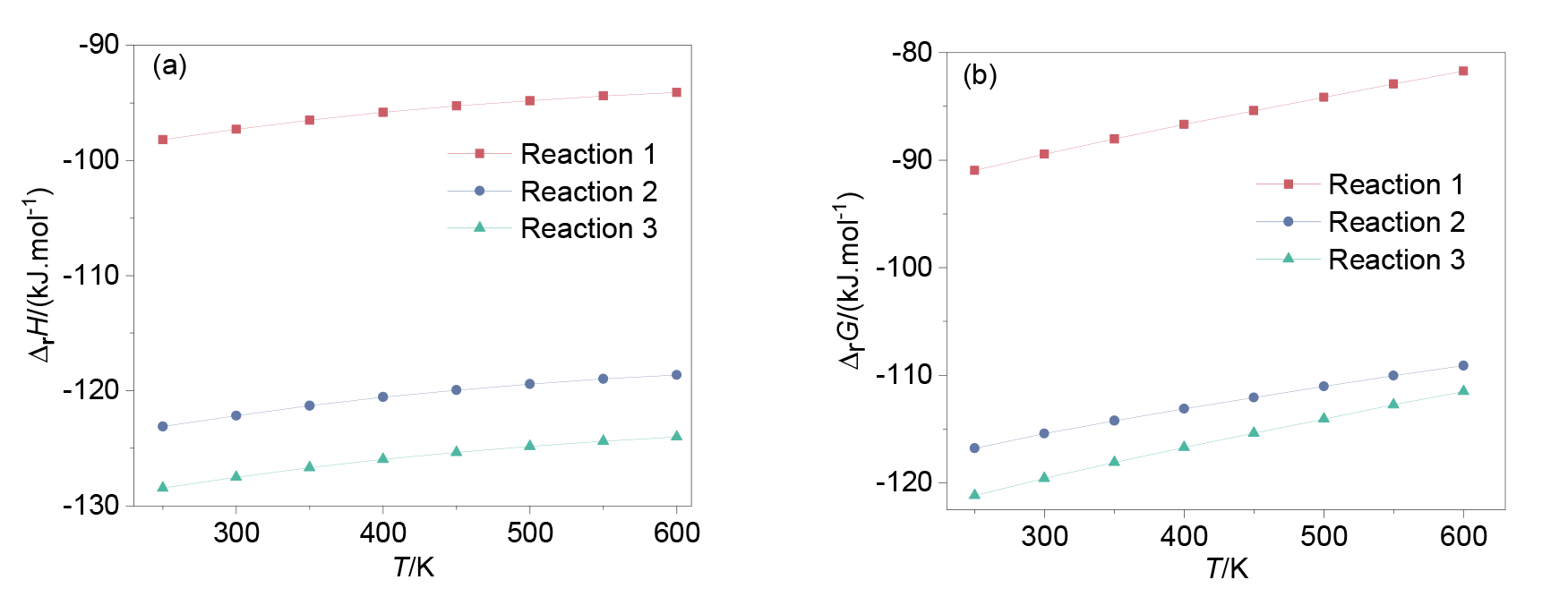
| Reaction | ΔrH/(kJ∙mol-1) | ΔS/(J∙mol-1∙K-1) | ΔrG/(kJ∙mol-1) |
|---|---|---|---|
| Reaction 1 | -97.3 | -26.2 | -89.5 |
| Reaction 2 | -122.2 | -22.5 | -115.5 |
| Reaction 3 | -127.6 | -26.8 | -119.6 |
| Reaction | ΔrH/(kJ∙mol-1) | ΔS/(J∙mol-1∙K-1) | ΔrG/(kJ∙mol-1) |
|---|---|---|---|
| Reaction 1 | -97.3 | -26.2 | -89.5 |
| Reaction 2 | -122.2 | -22.5 | -115.5 |
| Reaction 3 | -127.6 | -26.8 | -119.6 |

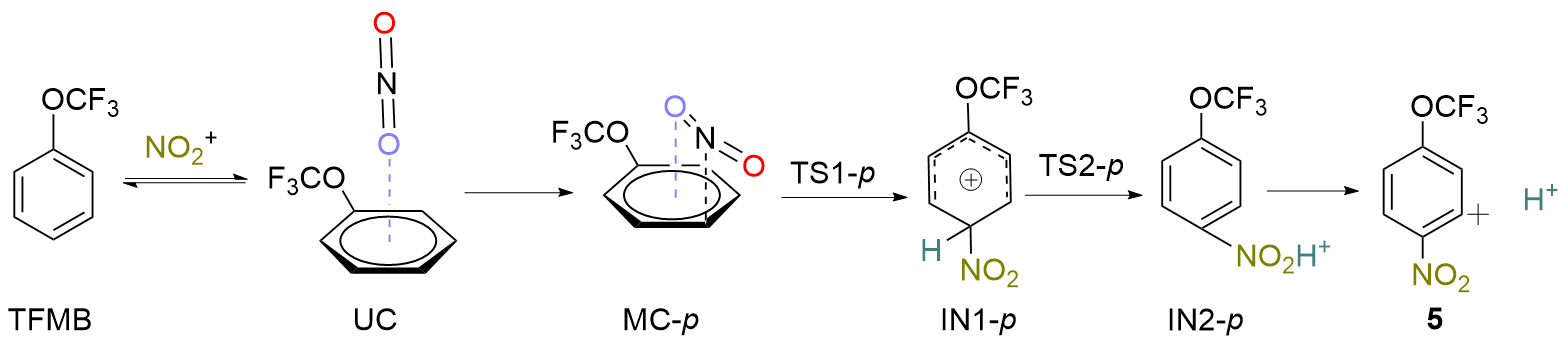
| Compd. | ΔH/(kJ∙mol-1) | ΔS/(J∙mol-1∙K-1) | ΔG/(kJ∙mol-1) |
|---|---|---|---|
| TFMB+ NO+ 2 | 0.0 | 0.0 | 0.0 |
| UC | -8.4 | -125.9 | 29.3 |
| MC-o | -7.9 | -138.9 | 33.9 |
| MC-m | -9.6 | -132.2 | 30.1 |
| MC-p | -9.6 | -131.4 | 29.7 |
| TS1-o | 7.9 | -162.3 | 56.5 |
| TS1-m | 15.5 | -158.6 | 62.8 |
| TS1-p | 1.7 | -149.8 | 46.4 |
| IN1-o | -45.2 | -156.5 | 1.3 |
| IN1-m | -18.4 | -156.1 | 28.0 |
| IN1-p | -69.0 | -159.0 | -21.8 |
| TS2-o | 64.4 | -160.2 | 112.5 |
| TS2-m | 75.3 | -154.0 | 120.9 |
| TS2-p | 48.1 | -159.8 | 95.8 |
| IN2-o | -125.1 | -160.7 | -77.0 |
| IN2-m | -140.2 | -161.5 | -92.0 |
| IN2-p | -154.0 | -161.5 | -105.9 |
| 3 | -113.4 | 10.0 | -116.3 |
| 4 | -123.0 | 10.5 | -127.6 |
| 5 | -114.6 | 10.5 | -117.6 |
| Compd. | ΔH/(kJ∙mol-1) | ΔS/(J∙mol-1∙K-1) | ΔG/(kJ∙mol-1) |
|---|---|---|---|
| TFMB+ NO+ 2 | 0.0 | 0.0 | 0.0 |
| UC | -8.4 | -125.9 | 29.3 |
| MC-o | -7.9 | -138.9 | 33.9 |
| MC-m | -9.6 | -132.2 | 30.1 |
| MC-p | -9.6 | -131.4 | 29.7 |
| TS1-o | 7.9 | -162.3 | 56.5 |
| TS1-m | 15.5 | -158.6 | 62.8 |
| TS1-p | 1.7 | -149.8 | 46.4 |
| IN1-o | -45.2 | -156.5 | 1.3 |
| IN1-m | -18.4 | -156.1 | 28.0 |
| IN1-p | -69.0 | -159.0 | -21.8 |
| TS2-o | 64.4 | -160.2 | 112.5 |
| TS2-m | 75.3 | -154.0 | 120.9 |
| TS2-p | 48.1 | -159.8 | 95.8 |
| IN2-o | -125.1 | -160.7 | -77.0 |
| IN2-m | -140.2 | -161.5 | -92.0 |
| IN2-p | -154.0 | -161.5 | -105.9 |
| 3 | -113.4 | 10.0 | -116.3 |
| 4 | -123.0 | 10.5 | -127.6 |
| 5 | -114.6 | 10.5 | -117.6 |
| [1] |
|
| [2] |
doi: 10.1016/j.cej.2016.09.034 |
| [3] |
pmid: 12696903 |
| [4] |
|
| [5] |
|
| [6] |
doi: 10.1016/j.psep.2021.11.036 |
| [7] |
|
|
(骆广生, 吕阳成, 王凯, 微化工技术, 北京, 化学工业出版社, 2020, pp. 1-14.)
|
|
| [8] |
doi: 10.1021/jp030167p |
| [9] |
doi: 10.1021/es500453g |
| [10] |
doi: 10.5958/2349-2988.2017.00048.1 |
| [11] |
doi: 10.1002/anie.v53.52 |
| [12] |
|
| [13] |
doi: 10.1002/anie.v54.47 |
| [14] |
doi: 10.1021/ja00291a015 |
| [15] |
pmid: 16872205 |
| [16] |
doi: 10.1007/s00894-017-3561-z |
| [17] |
|
| [18] |
doi: 10.1016/j.comptc.2021.113209 |
| [19] |
doi: 10.3866/PKU.WHXB20090332 |
|
(刘述斌, 物理化学学报, 2009, 25, 590.)
|
|
| [20] |
doi: 10.1021/cr990029p pmid: 12744694 |
| [21] |
|
| [22] |
doi: 10.1002/chem.v18.32 |
| [23] |
|
| [24] |
doi: 10.6023/cjoc202206033 |
|
(刘婷婷, 胡宇才, 沈安, 有机化学, 2023, 43, 622.)
doi: 10.6023/cjoc202206033 |
|
| [25] |
pmid: 27794598 |
| [26] |
doi: 10.1002/ijch.v33.4 |
| [27] |
doi: 10.1063/1.4963168 |
| [28] |
|
| [29] |
doi: 10.1016/j.molliq.2024.125097 |
| [30] |
doi: 10.5155/eurjchem.14.1.39-52.2340 |
| [31] |
|
| [32] |
doi: 10.1021/ct100326h pmid: 26616087 |
| [33] |
doi: 10.1002/jcc.v33.5 |
| [34] |
doi: 10.1063/5.0223879 |
| [35] |
|
| [36] |
doi: 10.3866/PKU.WHXB201401211 |
|
(付蓉, 卢天, 陈飞武, 物理化学学报, 2014, 30, 628.)
|
|
| [37] |
doi: 10.1039/d1cp02805g pmid: 34486612 |
| [38] |
doi: 10.3390/molecules21060748 |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||
