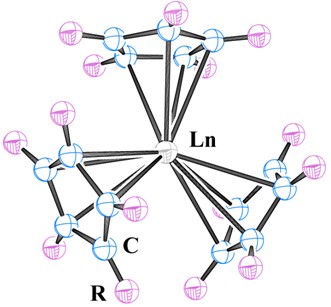
[1] Schumann, H.; Meesemarktscheffel, J. A.; Esser, L. Chem. Rev. 1995, 95, 865.[2] Wilkinson, G.; Birmingham, J. M. J. Am. Chem. Soc. 1954, 76, 6210. [3] Birmingham, J. M.; Wilkinson, G. J. Am. Chem. Soc. 1956, 78, 42. [4] Schaverien, C. J. Adv. Organomet. Chem. 1994, 36, 283. [5] Anwander, R.; Herrmann, W. A. Top. Curr. Chem. 1996, 179, 1. [6] Evans, W. J. Coord. Chem. Rev. 2000, 206, 263.  [7] Evans, W. J.; Gonzales, S. L.; Ziller, J. W. J. Am. Chem. Soc. 1991, 113, 7423. [8] Evans, W. J.; Forrestal, K. J.; Leman, J. T.; Ziller, J. W. Organometallics 1996, 15, 527. [9] Evans, W. J.; DeCoster, D. M.; Greaves, J. Macromolecules 1995, 28, 7929. [10] Evans, W. J.; Forrestal, K. J.; Ziller, J. W. J. Am. Chem. Soc. 1995, 117, 12635. [11] Evans, W. J.; Forrestal, K. J.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 9273. [12] Evans, W. J.; Forrestal, K. J.; Ansari, M. A.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 2180. [13] Evans, W. J.; Davis, B. L.; Champagne, T. M.; Ziller, J. W. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 12678. [14] Evans, W. J.; Davis, B. L.; Ziller, J. W. Inorg. Chem. 2001, 40, 6341. [15] Evans, W. J.; Perotti, J. M.; Kozimor, S. A.; Champagne, T. M.; Davis, B. L.; Nyce, G. W.; Fujimoto, C. H.; Clark, R. D.; Johnston, M. A.; Ziller, J. W. Organometallics 2005, 24, 3916. [16] Evans, W. J.; Seibel, C. A.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 6745. [17] Min, X.-M. Acta Chim. Sinica 1992, 50, 449. (闵新民, 化学学报, 1992, 50, 449.)[18] Min, X.-M. Acta Chim. Sinica 1994, 52, 996. (闵新民, 化学学报, 1994, 52, 996.)[19] Xu, Z.-F.; Xie, Y.-M.; Feng, W.-L.; Schaefer III, H. F. J. Phys. Chem. A 2003, 107, 2716. [20] Evans, W. J.; Mueller, T. J.; Ziller, J. W. J. Am. Chem. Soc. 2009, 131, 2678. [21] Ryan, M. F.; Eyler, J. R.; Richardson, D. E. J. Am. Chem. Soc. 1992, 114, 8611. [22] Sunderlint, L. S.; Armentrout, P. B. J. Am. Chem. Soc. 1989, 111, 3845. [23] Ephritikhine, M. Chem. Rev. 1997, 97, 2193. [24] Mueller, T. J.; Ziller, J. W.; Evans, W. J. Dalton Trans. 2010, 39, 6767. [25] Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297. [26] TURBOMOLE V6.3 2011, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989~2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com. [7] Evans, W. J.; Gonzales, S. L.; Ziller, J. W. J. Am. Chem. Soc. 1991, 113, 7423. [8] Evans, W. J.; Forrestal, K. J.; Leman, J. T.; Ziller, J. W. Organometallics 1996, 15, 527. [9] Evans, W. J.; DeCoster, D. M.; Greaves, J. Macromolecules 1995, 28, 7929. [10] Evans, W. J.; Forrestal, K. J.; Ziller, J. W. J. Am. Chem. Soc. 1995, 117, 12635. [11] Evans, W. J.; Forrestal, K. J.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 9273. [12] Evans, W. J.; Forrestal, K. J.; Ansari, M. A.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 2180. [13] Evans, W. J.; Davis, B. L.; Champagne, T. M.; Ziller, J. W. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 12678. [14] Evans, W. J.; Davis, B. L.; Ziller, J. W. Inorg. Chem. 2001, 40, 6341. [15] Evans, W. J.; Perotti, J. M.; Kozimor, S. A.; Champagne, T. M.; Davis, B. L.; Nyce, G. W.; Fujimoto, C. H.; Clark, R. D.; Johnston, M. A.; Ziller, J. W. Organometallics 2005, 24, 3916. [16] Evans, W. J.; Seibel, C. A.; Ziller, J. W. J. Am. Chem. Soc. 1998, 120, 6745. [17] Min, X.-M. Acta Chim. Sinica 1992, 50, 449. (闵新民, 化学学报, 1992, 50, 449.)[18] Min, X.-M. Acta Chim. Sinica 1994, 52, 996. (闵新民, 化学学报, 1994, 52, 996.)[19] Xu, Z.-F.; Xie, Y.-M.; Feng, W.-L.; Schaefer III, H. F. J. Phys. Chem. A 2003, 107, 2716. [20] Evans, W. J.; Mueller, T. J.; Ziller, J. W. J. Am. Chem. Soc. 2009, 131, 2678. [21] Ryan, M. F.; Eyler, J. R.; Richardson, D. E. J. Am. Chem. Soc. 1992, 114, 8611. [22] Sunderlint, L. S.; Armentrout, P. B. J. Am. Chem. Soc. 1989, 111, 3845. [23] Ephritikhine, M. Chem. Rev. 1997, 97, 2193. [24] Mueller, T. J.; Ziller, J. W.; Evans, W. J. Dalton Trans. 2010, 39, 6767. [25] Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297. [26] TURBOMOLE V6.3 2011, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989~2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com. |