化学学报 ›› 2023, Vol. 81 ›› Issue (6): 613-619.DOI: 10.6023/A23030071 上一篇 下一篇
研究论文
杨磊a, 葛娇阳a, 王访丽a, 吴汪洋a, 郑宗祥a, 曹洪涛a, 王洲c, 冉雪芹b,*( ), 解令海a,*()
), 解令海a,*()
投稿日期:2023-03-06
发布日期:2023-05-19
基金资助:
Lei Yanga, Jiaoyang Gea, Fangli Wanga, Wangyang Wua, Zongxiang Zhenga, Hongtao Caoa, Zhou Wangc, Xueqin Ranb(), Linhai Xiea()
Received:2023-03-06
Published:2023-05-19
Contact:
*E-mail: iamxqran@njtech.edu.cn; iamlhxie@njupt.edu.cn
Supported by:文章分享

有机半导体材料在有机发光二极管(OLED)、有机场效应晶体管(OFET)和有机太阳能电池(OSC)等领域应用广泛, 但有机半导体材料的迁移率较低, 不利于电子传输. 本工作基于芴分子设计并计算研究了具有“口”型结构的一类新型网格结构(OF), 与普通的大环分子相比, 它具有可以横向延伸的几何结构. 利用密度泛函理论研究了其分子结构, 环张力能和各种电子性质, 包括分子轨道、绝热电离势(IPa)、绝热电子亲和势(EAa)、重组能. 此外, 利用非共价相互作用和Normal Mode (NM)分析方法分别研究了分子内的弱相互作用和每个振动模式对OF重组能的贡献. 结果表明OF分子具有非常弱的环张力能(8.20 kJ/mol), 比较容易在实验室中合成. 通过分子内弱相互作用分析发现, 由于cis-OF横梁上的两个芴单元距离较近并且产生了一定的角度, 从而产生了π-π相互作用. 与Bis-Fl1, Bis-Fl2, Quarter-Fl1和Quarter-Fl2相比, OF分子的IPa降低, EAa提升, 证明了格子化效应能提高分子的空穴和电子注入能力. 同时, OF具有较低的IPa, 是一种非常具有潜力的p型分子材料. 另外, 当形成口字型芴格时, OF的空穴重组能和电子重组能都有所降低, 说明格子化效应是有效降低重组能的一种方式, 为未来设计具有优秀电荷传输性质的有机半导体材料提供了一种有效的策略.
杨磊, 葛娇阳, 王访丽, 吴汪洋, 郑宗祥, 曹洪涛, 王洲, 冉雪芹, 解令海. 一种基于芴的大环结构的有效降低内重组能的理论研究[J]. 化学学报, 2023, 81(6): 613-619.
Lei Yang, Jiaoyang Ge, Fangli Wang, Wangyang Wu, Zongxiang Zheng, Hongtao Cao, Zhou Wang, Xueqin Ran, Linhai Xie. A Theoretical Study on the Effective Reduction of Internal Reorganization Energy Based on the Macrocyclic Structure of Fluorene[J]. Acta Chimica Sinica, 2023, 81(6): 613-619.
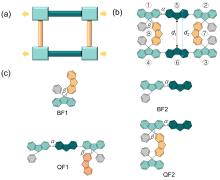

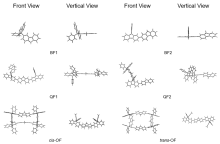
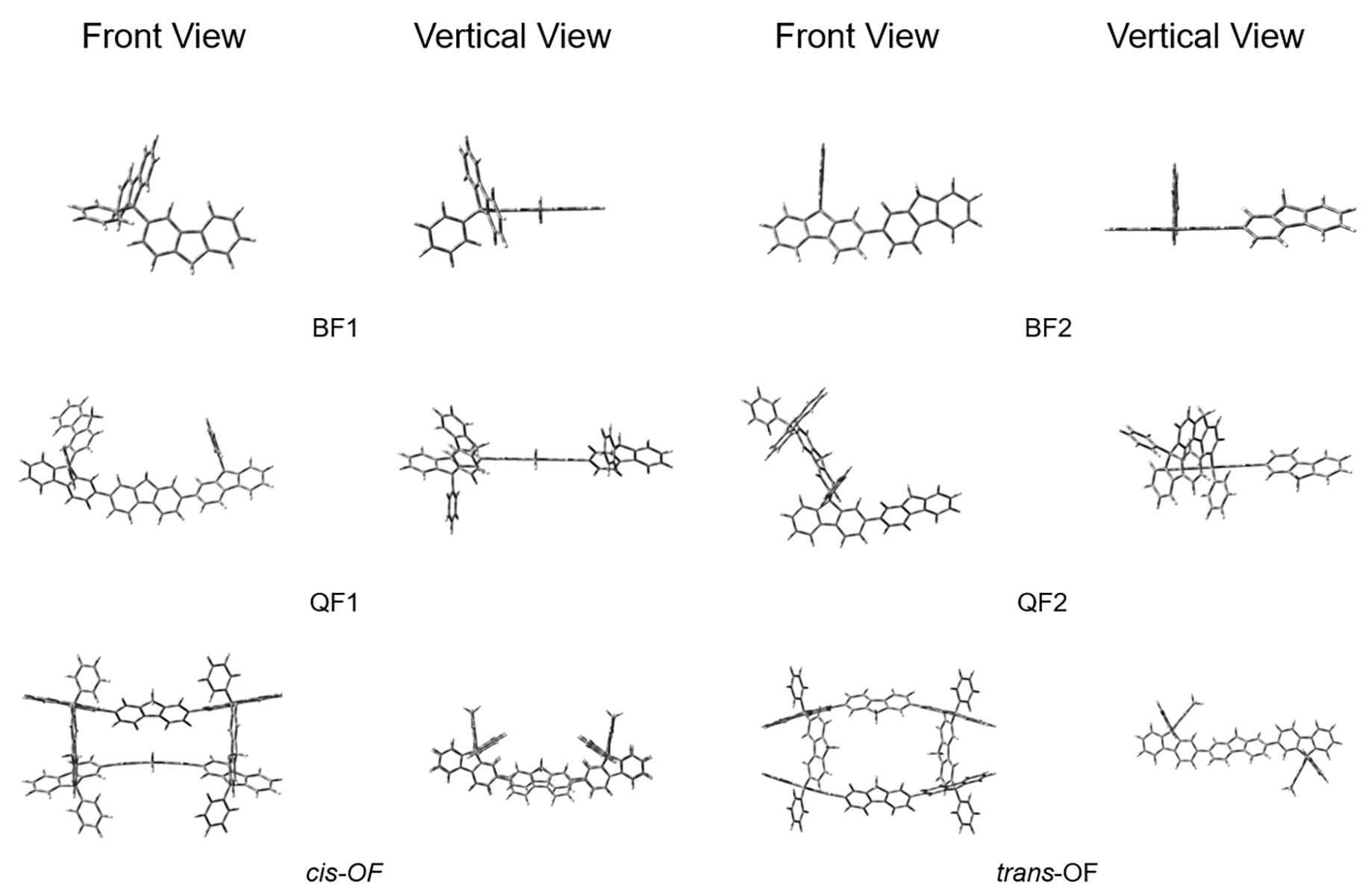
| 分子名称 | α | β |
|---|---|---|
| BF1 | -84.50 | |
| BF2 | 37.33 | |
| QF1 | -36.73, -36.98 | -84.77 |
| QF2 | 37.10 | 86.13, -85.65 |
| cis-OF | -40.58, 40.58, -34.85, 34.85 | -26.25, 26.25, 28.48, -28.48 |
| trans-OF | 88.76, 88.38, -88.76, -88.38 | -35.60, 41.70, 35.60, -41.70 |
| 分子名称 | α | β |
|---|---|---|
| BF1 | -84.50 | |
| BF2 | 37.33 | |
| QF1 | -36.73, -36.98 | -84.77 |
| QF2 | 37.10 | 86.13, -85.65 |
| cis-OF | -40.58, 40.58, -34.85, 34.85 | -26.25, 26.25, 28.48, -28.48 |
| trans-OF | 88.76, 88.38, -88.76, -88.38 | -35.60, 41.70, 35.60, -41.70 |


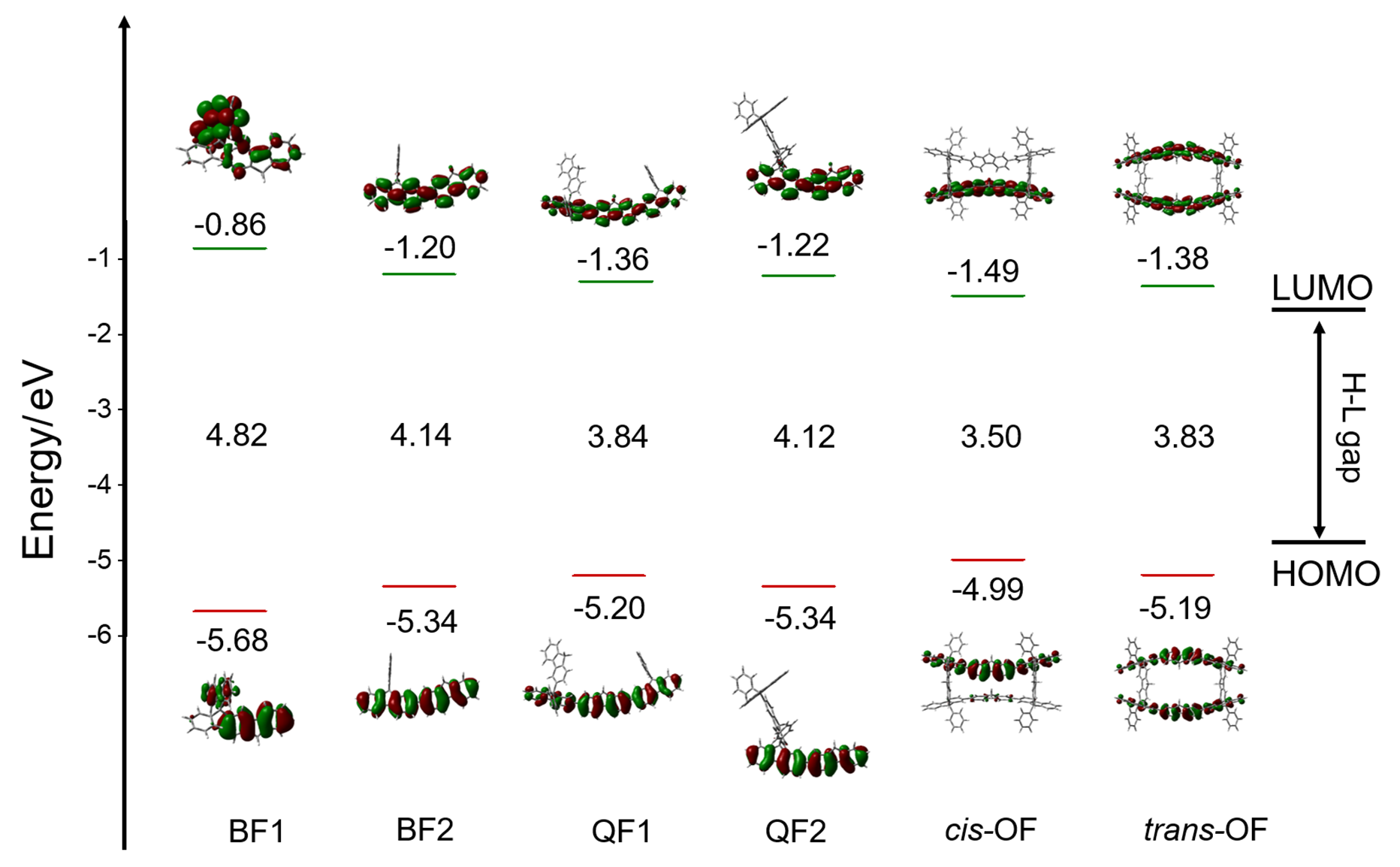


| 分子名称 | λ(h) | IPa | RMSD(VS+) | λ(e) | EAa | RMSD(VS-) |
|---|---|---|---|---|---|---|
| BF1 | 0.146 | 6.856 | 0.104 | 0.177 | -0.287 | 0.138 |
| BF2 | 0.255 | 6.428 | 0.202 | 0.355 | 0.180 | 0.369 |
| QF1 | 0.203 | 6.073 | 0.395 | 0.276 | 0.574 | 0.684 |
| QF2 | 0.153 | 6.280 | 0.185 | 0.216 | 0.324 | 0.484 |
| cis-OF | 0.128 | 5.829 | 0.140 | 0.148 | 0.708 | 0.121 |
| trans-OF | 0.121 | 5.848 | 0.093 | 0.145 | 0.735 | 0.115 |
| 分子名称 | λ(h) | IPa | RMSD(VS+) | λ(e) | EAa | RMSD(VS-) |
|---|---|---|---|---|---|---|
| BF1 | 0.146 | 6.856 | 0.104 | 0.177 | -0.287 | 0.138 |
| BF2 | 0.255 | 6.428 | 0.202 | 0.355 | 0.180 | 0.369 |
| QF1 | 0.203 | 6.073 | 0.395 | 0.276 | 0.574 | 0.684 |
| QF2 | 0.153 | 6.280 | 0.185 | 0.216 | 0.324 | 0.484 |
| cis-OF | 0.128 | 5.829 | 0.140 | 0.148 | 0.708 | 0.121 |
| trans-OF | 0.121 | 5.848 | 0.093 | 0.145 | 0.735 | 0.115 |


| [1] |
Yan, C.; Qin, J.; Wang, Y.; Li, G.; Cheng, P. Adv. Energy Mater. 2022, 12, 2201087.
doi: 10.1002/aenm.v12.26 |
| [2] |
Reissig, L.; Dalgleish, S.; Awaga, K. Sci. Rep. 2018, 8, 15415.
doi: 10.1038/s41598-018-33822-z pmid: 30337667 |
| [3] |
Li, M.; Lv, A. F. Chin. J. Org. Chem. 2022, 42, 54. (in Chinese)
doi: 10.6023/cjoc202107016 |
|
(李敏, 吕爱风, 有机化学, 2022, 42, 54.)
doi: 10.6023/cjoc202107016 |
|
| [4] |
Zhou, M.; Li, J.; Cheng, J.; Ge, C. W.; Cheng, T. Y.; Gao, X. K. Chin. J. Org. Chem. 2021, 41, 4400. (in Chinese)
doi: 10.6023/cjoc202105023 |
|
(周敏, 李晶, 程杰, 葛从伍, 程探宇, 高希珂, 有机化学, 2021, 41, 4400.)
doi: 10.6023/cjoc202105023 |
|
| [5] |
Li, X.; Wang, Z.; Chen, K.; Zemlyanov, D.Y.; You, L.; Mei, J. ACS Appl. Mater. Interfaces 2021, 13, 5312.
doi: 10.1021/acsami.0c19685 |
| [6] |
Cai, G.; Cui, P.; Shi, W.; Morris, S.; Lou, S. N.; Chen, J.; Ciou, J. H.; Paidi, V. K.; Lee, K. S.; Li, S.; Lee, P. S. Adv. Sci. 2020, 7, 1903109.
doi: 10.1002/advs.v7.20 |
| [7] |
Liu, B.; Bao, Y.; Ling, H.-F.; Zhu, W.-S.; Gong, R.-J.; Lin, J.-Y.; Xie, L.-H.; Yi, M.-D.; Huang, W. Chinese J. Polym. Sci. 2016, 34, 1183.
doi: 10.1007/s10118-016-1826-0 |
| [8] |
Li, T. F.; Zhan, X. W. Acta Chim. Sinica 2021, 79, 257. (in Chinese)
doi: 10.6023/A20110502 |
|
(李腾飞, 占肖卫, 化学学报, 2021, 79, 257.)
doi: 10.6023/A20110502 |
|
| [9] |
Zhu, K.; Tang, D.; Zhang, K.; Wang, Z.; Ding, L.; Liu, Y.; Yuan, L.; Fan, J.; Song, B.; Zhou, Y.; Li, Y. Org. Electron. 2017, 48, 179
doi: 10.1016/j.orgel.2017.06.009 |
| [10] |
Brebels, J.; Kesters, J.; Defour, M.; Pirotten, G.; Van Mele, B.; Manca, J.; Lutsen, L.; Vanderzande, D.; Maes, W. Polymer. 2018. 137, 303.
doi: 10.1016/j.polymer.2018.01.027 |
| [11] |
Feng, Q.; Xie, S.; Tan, K.; Zheng, X.; Yu, Z.; Li, L.; Liu, B.; Li, B.; Yu, M.; Yu, Y.; Zhang, X.; Xie, L.; Huang, W. ACS Appl. Polym. Mater. 2019, 1, 2441.
doi: 10.1021/acsapm.9b00559 |
| [12] |
Yang, S.; Streater, D.; Fiankor, C.; Zhang, J.; Huang, J. J. Am. Chem. Soc. 2021, 143, 1061.
doi: 10.1021/jacs.0c11719 |
| [13] |
Schober, C.; Reuter, K.; Oberhofer, H. J. Phys. Chem. Lett. 2016, 7, 3973.
doi: 10.1021/acs.jpclett.6b01657 |
| [14] |
Friederich, P.; Gómez, V.; Sprau, C.; Meded, V.; Strunk, T.; Jenne, M.; Magri, A.; Symalla, F.; Colsmann, A.; Ruben, M.; Wenzel, W. Adv. Mater. 2017, 29, 1703505.
doi: 10.1002/adma.201703505 |
| [15] |
Chen, J.; Zhang, W.; Wang, L.; Yu, G. Adv. Mater. 2023, 35, 2210772.
doi: 10.1002/adma.v35.11 |
| [16] |
Marcus, R. A. Annu. Rev. Phys. Chem. 2003, 15, 155.
doi: 10.1146/physchem.1964.15.issue-1 |
| [17] |
Xie, X.; Wei, Y.; Lin, D.; Zhong, C.; Xie, L.; Huang, W. Chinese J. Chem. 2019, 38, 103.
doi: 10.1002/cjoc.v38.1 |
| [18] |
Yang, L.; Mao, J.; Yin, C.-Z.; Akbar Ali, M.; Wu, X.-P.; Dong, C.-Y.; Liu, Y.-Y.; Wei, Y.; Xie, L.-H.; Ran, X.-Q.; Huang, W. New J. Chem. 2019, 43, 7790.
doi: 10.1039/c9nj00482c |
| [19] |
Yang, L.; Yin, C. Z.; Ali, M. A.; Dong, C. Y.; Xie, X. M.; Wu, X. P.; Mao, J.; Wang, Y. X.; Yu, Y.; Xie, L. H.; Bian, L. Y.; Bao, J. M.; Ran, X. Q.; Huang, W. Chinese J. Chem. 2019, 37, 915.
doi: 10.1002/cjoc.201900229 |
| [20] |
Yu, M.-N.; Ou, C.-J.; Liu, B.; Lin, D.-Q.; Liu, Y.-Y.; Xue, W.; Lin, Z.-Q.; Lin, J.-Y.; Qian, Y.; Wang, S.-S.; Cao, H.-T.; Bian, L.-Y.; Xie, L.-H.; Huang, W. J. Polym. Sci. B Polym. Phys. 2017, 35, 155.
|
| [21] |
Sedghi, G.; Esdaile, L. J.; Anderson, H. L.; Martin, S.; Bethell, D.; Higgins, S. J.; Nichols, R. J., Adv. Mater. 2012, 24, 653.
doi: 10.1002/adma.201103109 |
| [22] |
Lin, D.; Zhang, W.; Yin, H.; Hu, H.; Li, Y.; Zhang, H.; Wang, L.; Xie, X.; Hu, H.; Yan, Y.; Ling, H.; Liu, J.; Qian, Y.; Tang, L.; Wang, Y.; Dong, C.; Xie, L.; Zhang, H.; Wang, S.; Wei, Y.; Guo, X.; Lu, D.; Huang, W. Research 2022, 2022, 9820585.
|
| [23] |
Chang, A. C.; Messikh, M. B.; Kaiser, M.; Carter, K. R. ACS Appl. Polym. Mater. 2021, 3, 3595.
doi: 10.1021/acsapm.1c00489 |
| [24] |
Han, Y.; Bai, L.; Lin, J.; Ding, X.; Xie, L.; Huang, W. Adv. Funct. 2021, 31, 2105092.
|
| [25] |
Liu, A.; Liu, Z.; Lin, H.; Tang, W.; Lin, Z.; Zhang, W.; Qi, Z.; Gu, X.; Mo, Y.; Hou, L. J. Mater. Chem. C 2020, 8, 9303.
doi: 10.1039/D0TC01952F |
| [26] |
Nakahama, T.; Kitagawa, D.; Sotome, H.; Ito, S.; Miyasaka, H.; Kobatake, S. J. Phys. Chem. C 2017, 121, 6272.
doi: 10.1021/acs.jpcc.6b12819 |
| [27] |
Liu, J.-F.; Wang, X.-Q.; Yu, Y.-J.; Zou, S.-N.; Yang, S.-Y.; Jiang, Z.-Q.; Liao, L.-S. Org. Electron. 2021, 91, 106088.
doi: 10.1016/j.orgel.2021.106088 |
| [28] |
Wheeler, S. E.; Houk, K. N.; Schleyer, P. v. R.; Allen, W. D. J. Am. Chem. Soc. 2009, 131, 2547.
doi: 10.1021/ja805843n pmid: 19182999 |
| [29] |
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B. S., G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, C.1, Gaussian, Inc., Wallingford, CT, 2009.
|
| [30] |
Li, Y.; Wang, Z.; Cai, X.; Liu, K.; Dong, J.; Chang, S.; Su, S.-J. Dyes Pigm. 2019, 163, 249.
doi: 10.1016/j.dyepig.2018.12.001 |
| [31] |
Lu, T.; Chen, F. J. Comput. Chem. 2022, 43, 8.
|
| [32] |
Johnson, E. R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A. J.; Yang, W. J. Am. Chem. Soc. 2010, 132, 6498.
doi: 10.1021/ja100936w pmid: 20394428 |
| [33] |
Reimers, J. R. J. Chem. Phys. 2001, 115, 9103.
doi: 10.1063/1.1412875 |
| [1] | 黄广龙, 薛小松. “陈试剂”作为三氟甲基源机理的理论研究[J]. 化学学报, 2024, 82(2): 132-137. |
| [2] | 葛凤洁, 张开志, 曹清鹏, 徐慧, 周涛, 张文浩, 班鑫鑫, 张晓波, 李娜, 朱鹏. 柔性芴基嵌段型延迟荧光二聚体的设计、合成及电致发光性能[J]. 化学学报, 2023, 81(9): 1157-1166. |
| [3] | 梁雪峰, 荆剑, 冯昕, 赵勇泽, 唐新员, 何燕, 张立胜, 李慧芳. 共价有机框架COF66/COF366的电子结构: 从单体到二维平面聚合物[J]. 化学学报, 2023, 81(7): 717-724. |
| [4] | 张少秦, 李美清, 周中军, 曲泽星. 多共振热激活延迟荧光过程的理论研究[J]. 化学学报, 2023, 81(2): 124-130. |
| [5] | 王娟, 肖华敏, 谢丁, 郭元茹, 潘清江. 铜掺杂与氮化碳复合氧化锌材料结构和二氧化氮气体传感性质的密度泛函理论计算[J]. 化学学报, 2023, 81(11): 1493-1499. |
| [6] | 刘金晶, 杨娜, 李莉, 魏子栋. 铂活性位空间结构调控氧还原机理的理论研究★[J]. 化学学报, 2023, 81(11): 1478-1485. |
| [7] | 刘玉玉, 陈捷锋, 邵振, 魏颖, 凌海峰, 解令海. 基于H型芴基小分子的双极性有机场效应晶体管存储器[J]. 化学学报, 2023, 81(11): 1508-1514. |
| [8] | 栾雪菲, 王聪芝, 夏良树, 石伟群. 铀酰与羧酸和肟基类配体相互作用的理论研究[J]. 化学学报, 2022, 80(6): 708-713. |
| [9] | 王珞聪, 李哲伟, 岳彩巍, 张培焕, 雷鸣, 蒲敏. 电场下偶氮苯衍生物分子顺反异构化反应机理的理论研究[J]. 化学学报, 2022, 80(6): 781-787. |
| [10] | 曹洪涛, 侯鹏飞, 曹庆, 李延昂, 汪莎莎, 解令海. 基于氰基化9-苯基芴衍生物的激基复合物发光与性质研究[J]. 化学学报, 2022, 80(11): 1476-1484. |
| [11] | 王英辉, 魏思敏, 段金伟, 王康. 理论研究“受阻路易斯酸碱对”催化的烯醇硅醚氢化反应机理[J]. 化学学报, 2021, 79(9): 1164-1172. |
| [12] | 熊昆, 陈伽瑶, 杨娜, 蒋尚坤, 李莉, 魏子栋. 理论探究水溶液条件对TMNxCy催化氮还原性能的增强机制[J]. 化学学报, 2021, 79(9): 1138-1145. |
| [13] | 胡鑫明, 钟春晓, 李晓艳, 贾雄, 魏颖, 解令海. 环戊并二噻吩衍生物的合成及其应用[J]. 化学学报, 2021, 79(8): 953-966. |
| [14] | 满清敏, 付尊蕴, 刘甜甜, 郑明月, 蒋华良. Cu催化偶联反应合成烷基芳基醚的DFT机理研究[J]. 化学学报, 2021, 79(7): 948-952. |
| [15] | 王岩, 田英齐, 金钟, 索兵兵. 基于GPU的Hartree-Fock与密度泛函算法及程序[J]. 化学学报, 2021, 79(5): 653-657. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
