化学学报 ›› 2026, Vol. 84 ›› Issue (6): 828-839.DOI: 10.6023/A26040105 上一篇 下一篇
研究论文
马玲玲a,b, 凌琳b, 李玉学b,*( ), 吕龙b,*()
), 吕龙b,*()
投稿日期:2026-04-04
发布日期:2026-04-27
基金资助:
Lingling Maa,b, Lin Lingb, Yuxue Lib,*(), Long Lub,*()
Received:2026-04-04
Published:2026-04-27
Contact:
E-mail: Supported by:文章分享
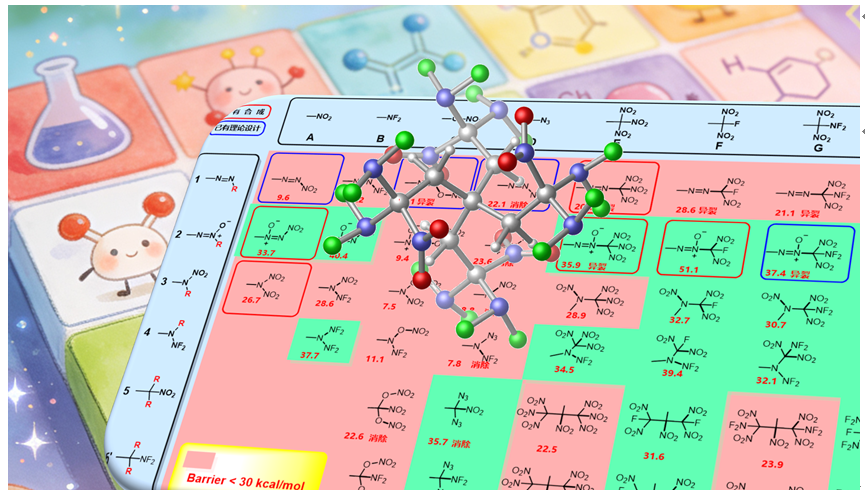
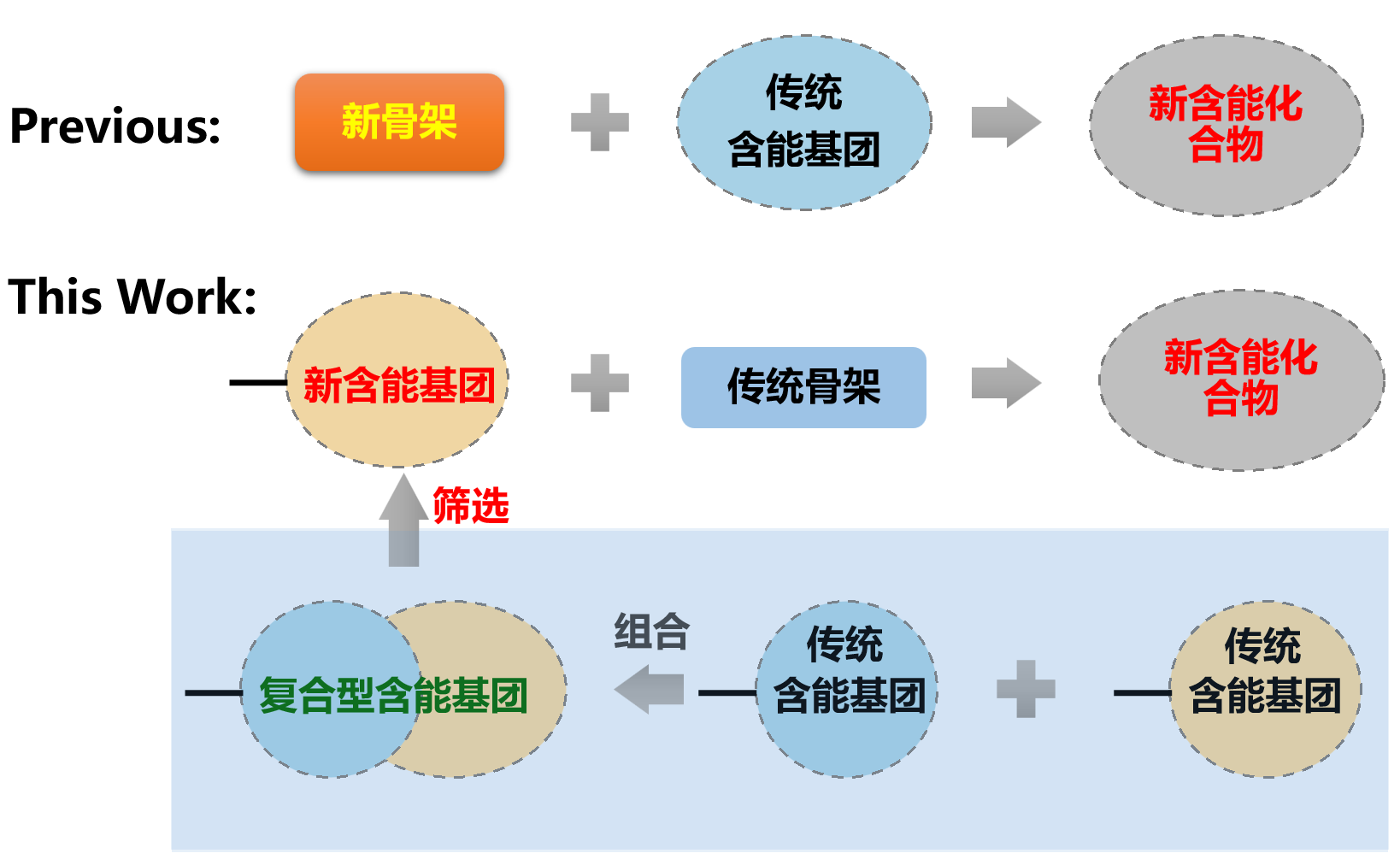
打破含能材料的发展瓶颈, 是近年来亟待攻克的难题. 本文尝试从设计新型含能基团的角度来探索新含能分子. 通过将传统含能基团进行组合, 使用密度泛函理论(DFT)方法计算了分子结构、初始热分解机理, 使用KJ方程和EXPLO5计算了爆轰性能, 筛选出39个含能基团, 初始分解能垒≥30 kcal•mol-1. 其中5个已有实验合成的结构在热稳定性方面, 计算结果和实验十分吻合. 尝试用烷烃和多胺骨架进行分子设计, 所得分子密度>2.0 g•cm-3, 爆速>8000 m•s-1, 爆压>33 GPa. 分子内张力/位阻、以及N原子孤对电子的超共轭效应, 对最终含能分子的动力学稳定性具有重要影响.
马玲玲, 凌琳, 李玉学, 吕龙. 复合型含能基团结构和性能的理论研究[J]. 化学学报, 2026, 84(6): 828-839.
Lingling Ma, Lin Ling, Yuxue Li, Long Lu. Theoretical Study on the Structure and Properties of Composite Energetic Groups[J]. Acta Chimica Sinica, 2026, 84(6): 828-839.
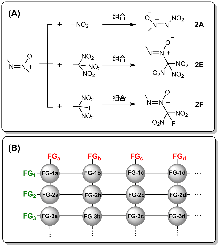
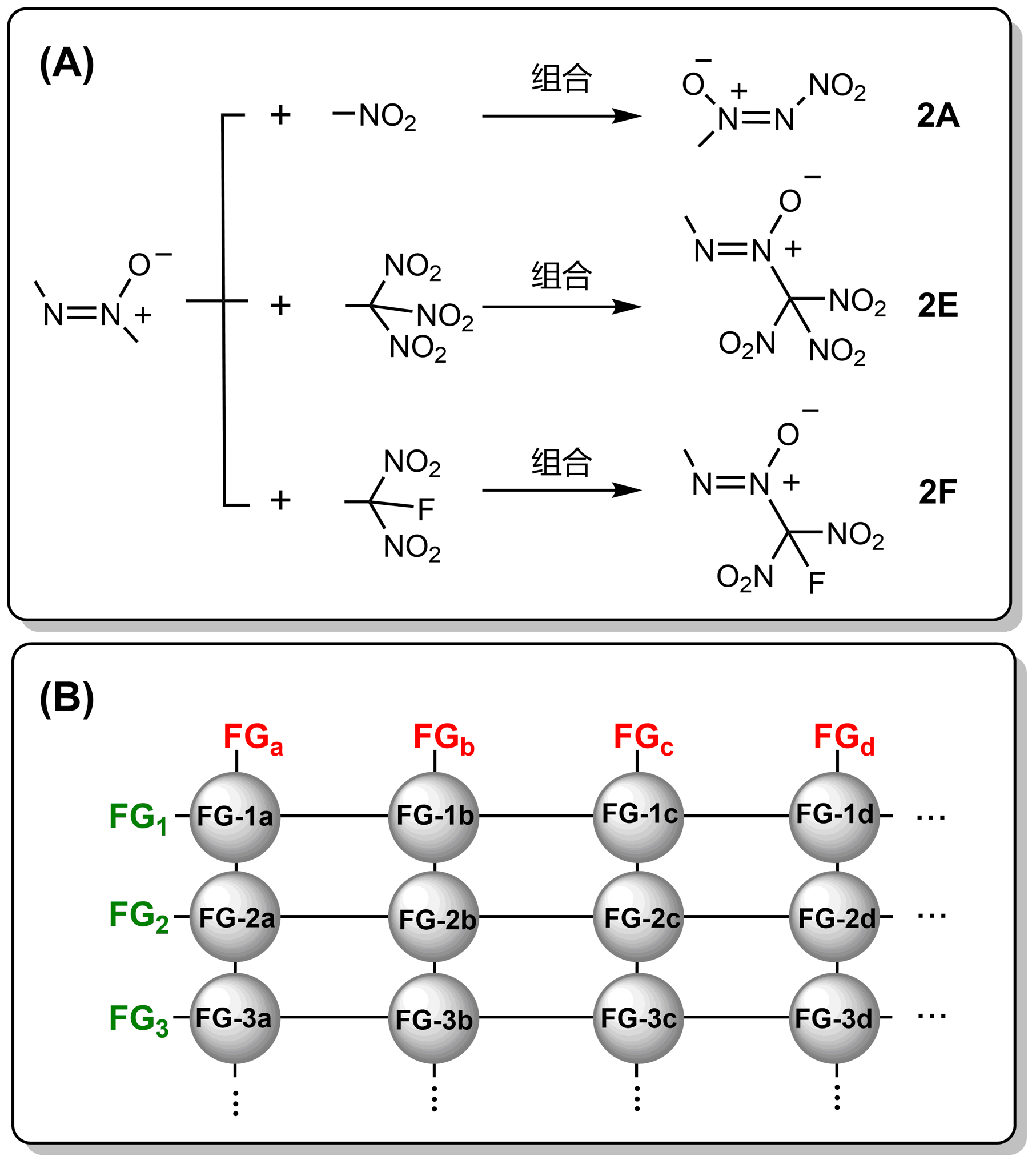
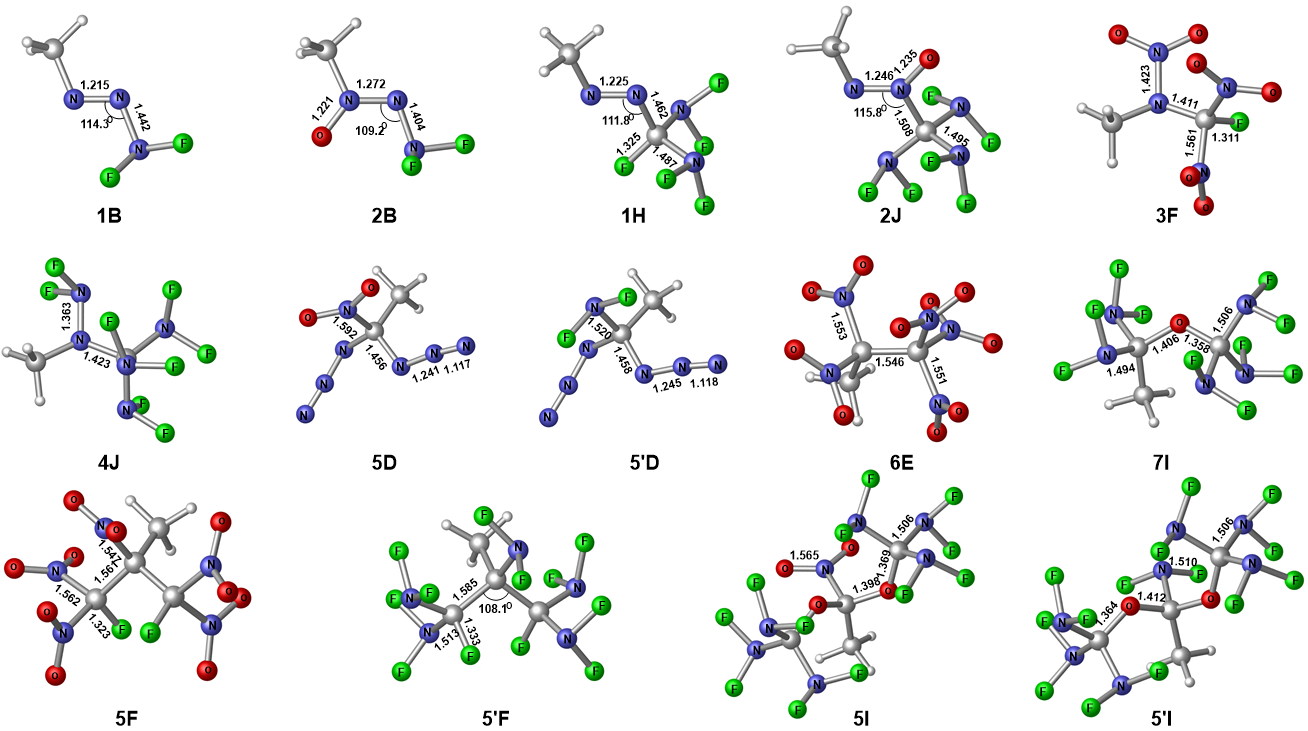
| Compd. | Barrier | ∆fHSolid | ρ | Q/(cal•g-1) | D/(km•s-1) | P/GPa | Compd. | Barrier | ∆fHSolid | ρ | Q/(cal•g-1) | D/(km•s-1) | P/GPa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1H | 39.2 | -156.2 | 1.94 | 1516 | 9.053 | 36.8 | 5I | 40.2 | -619.9 | 2.30 | 1617 | 10.098 | 50.6 |
| 2A | 33.7 | 107.1 | 1.64 | 1644 | 8.881 | 33.0 | 5'D | 33.9 | 524.6 | 1.65 | 1571 | 8.412 | 28.6 |
| 2B | 40.4 | 52.4 | 1.70 | 1670 | 9.165 | 34.8 | 5'F | 32.8 | -509.3 | 2.09 | 1486 | 9.692 | 44.2 |
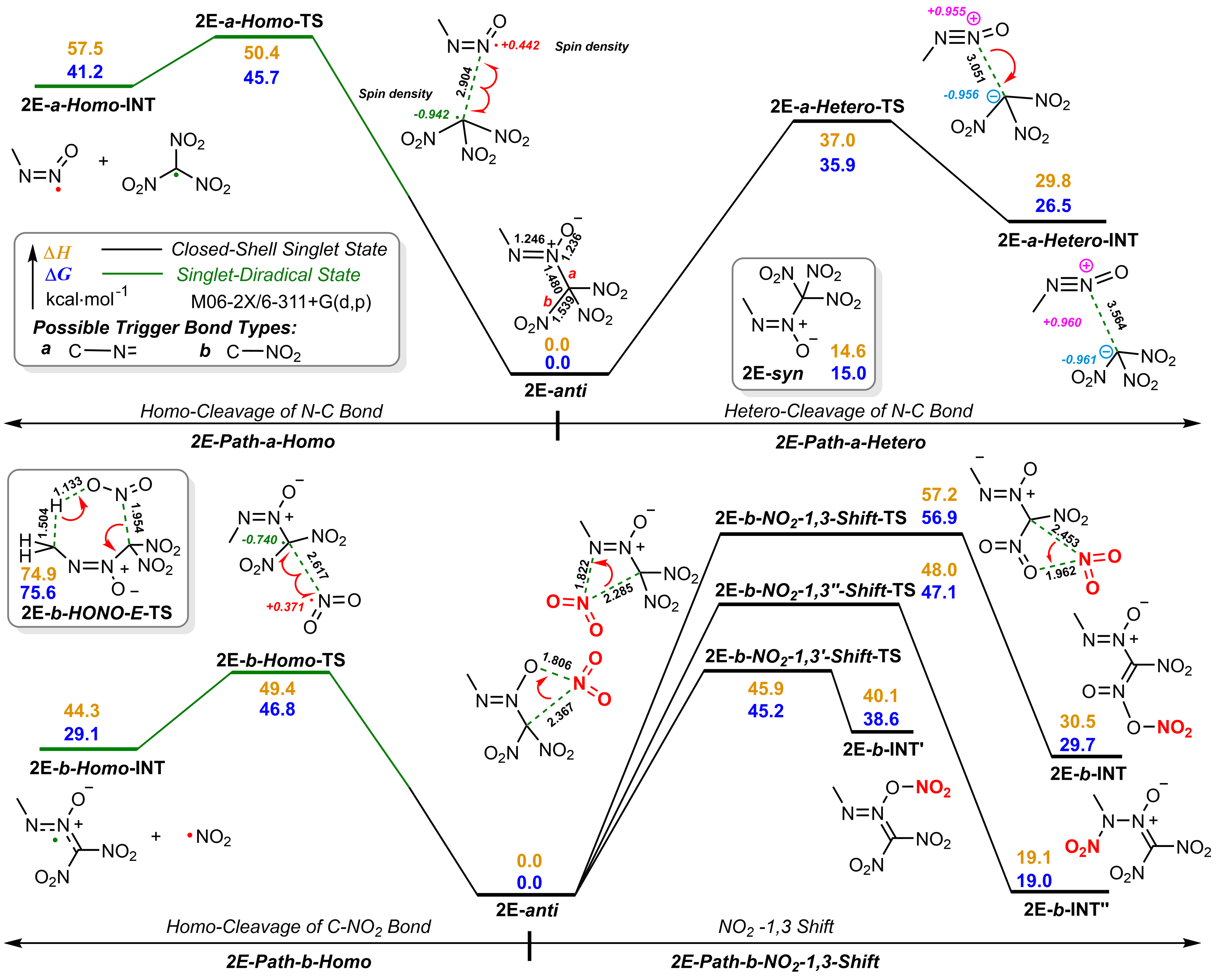
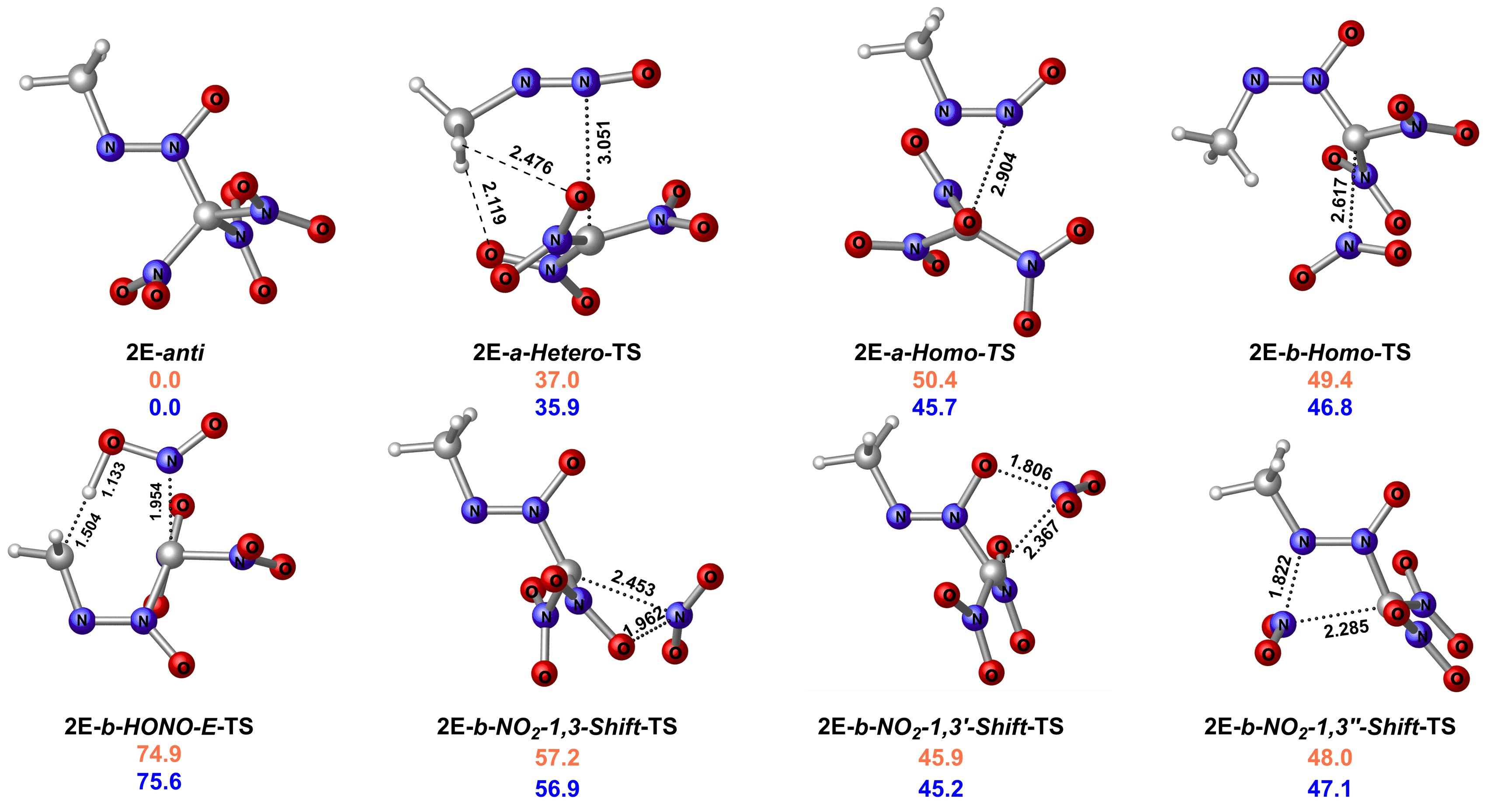
| 2E | 35.9 | 136.5 | 1.88 | 1472 | 9.134 | 38.0 | 5'H | 36.5 | -641.6 | 2.35 | 1556 | 9.776 | 47.9 |
| 2F | 51.1 | -108.6 | 1.80 | 1410 | 8.800 | 33.2 | 5'I | 39.2 | -646.8 | 2.34 | 1649 | 10.136 | 51.5 |
| 2G | 37.4 | 76.8 | 1.89 | 1703 | 9.606 | 41.0 | 6D | 30.7 | 111.7 | 1.74 | 1243 | 8.578 | 31.9 |
| 2H | 47.9 | -200.3 | 1.98 | 1473 | 9.512 | 41.4 | 6E | 32.2 | -90.0 | 2.00 | 1292 | 9.134 | 39.4 |
| 2J | 39.1 | 4.0 | 2.07 | 1719 | 10.107 | 47.9 | 6F | 34.7 | -316.4 | 1.94 | 1363 | 9.073 | 37.0 |
| 3F | 32.7 | -208.5 | 1.90 | 1321 | 8.982 | 35.8 | 6G | 30.0 | -127.6 | 2.02 | 1496 | 9.638 | 43.0 |
| 3G | 30.7 | -22.0 | 1.97 | 1483 | 9.566 | 41.7 | 6H | 33.7 | -388.4 | 2.09 | 1504 | 9.845 | 45.7 |
| 3H | 33.2 | -267.2 | 2.06 | 1509 | 9.900 | 45.9 | 6I | 37.6 | -370.8 | 2.12 | 1604 | 9.895 | 46.5 |
| 4B | 37.7 | -49.5 | 1.91 | 1142 | 6.618 | 19.0 | 6J | 30.7 | -177.0 | 2.15 | 1737 | 10.268 | 50.6 |
| 4E | 34.5 | -26.1 | 1.95 | 1479 | 9.475 | 40.6 | 7A | 38.8 | -210.1 | 1.96 | 1495 | 9.762 | 43.3 |
| 4F | 39.4 | -264.7 | 1.95 | 1573 | 9.663 | 42.2 | 7D | 32.2 | 151.0 | 1.88 | 1663 | 9.172 | 37.1 |
| 4G | 32.1 | -79.6 | 2.02 | 1677 | 9.964 | 45.9 | 7F | 35.9 | -380.6 | 2.09 | 1511 | 9.852 | 45.7 |
| 4H | 43.6 | -335.0 | 2.12 | 1567 | 9.614 | 43.9 | 7G | 31.8 | -183.1 | 2.17 | 1732 | 10.325 | 51.4 |
| 4J | 37.7 | -134.6 | 2.19 | 1775 | 10.133 | 49.7 | 7H | 39.6 | -444.7 | 2.01 | 1592 | 8.956 | 36.8 |
| 5D | 35.7 | 543.0 | 1.63 | 1464 | 8.174 | 27.9 | 7I | 40.1 | -435.1 | 2.20 | 1612 | 9.932 | 47.9 |
| 5F | 31.6 | -491.2 | 2.03 | 1312 | 9.248 | 39.5 | 7J | 33.5 | -249.0 | 2.27 | 1758 | 10.075 | 50.1 |
| 5H | 32.0 | -626.1 | 2.28 | 1424 | 9.956 | 49.0 | CL-20 | 35.8 | 466.5 | 2.07 | 1618 | 9.796 | 46.1 |
| Compd. | Barrier | ∆fHSolid | ρ | Q/(cal•g-1) | D/(km•s-1) | P/GPa | Compd. | Barrier | ∆fHSolid | ρ | Q/(cal•g-1) | D/(km•s-1) | P/GPa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1H | 39.2 | -156.2 | 1.94 | 1516 | 9.053 | 36.8 | 5I | 40.2 | -619.9 | 2.30 | 1617 | 10.098 | 50.6 |
| 2A | 33.7 | 107.1 | 1.64 | 1644 | 8.881 | 33.0 | 5'D | 33.9 | 524.6 | 1.65 | 1571 | 8.412 | 28.6 |
| 2B | 40.4 | 52.4 | 1.70 | 1670 | 9.165 | 34.8 | 5'F | 32.8 | -509.3 | 2.09 | 1486 | 9.692 | 44.2 |
| 2E | 35.9 | 136.5 | 1.88 | 1472 | 9.134 | 38.0 | 5'H | 36.5 | -641.6 | 2.35 | 1556 | 9.776 | 47.9 |
| 2F | 51.1 | -108.6 | 1.80 | 1410 | 8.800 | 33.2 | 5'I | 39.2 | -646.8 | 2.34 | 1649 | 10.136 | 51.5 |
| 2G | 37.4 | 76.8 | 1.89 | 1703 | 9.606 | 41.0 | 6D | 30.7 | 111.7 | 1.74 | 1243 | 8.578 | 31.9 |
| 2H | 47.9 | -200.3 | 1.98 | 1473 | 9.512 | 41.4 | 6E | 32.2 | -90.0 | 2.00 | 1292 | 9.134 | 39.4 |
| 2J | 39.1 | 4.0 | 2.07 | 1719 | 10.107 | 47.9 | 6F | 34.7 | -316.4 | 1.94 | 1363 | 9.073 | 37.0 |
| 3F | 32.7 | -208.5 | 1.90 | 1321 | 8.982 | 35.8 | 6G | 30.0 | -127.6 | 2.02 | 1496 | 9.638 | 43.0 |
| 3G | 30.7 | -22.0 | 1.97 | 1483 | 9.566 | 41.7 | 6H | 33.7 | -388.4 | 2.09 | 1504 | 9.845 | 45.7 |
| 3H | 33.2 | -267.2 | 2.06 | 1509 | 9.900 | 45.9 | 6I | 37.6 | -370.8 | 2.12 | 1604 | 9.895 | 46.5 |
| 4B | 37.7 | -49.5 | 1.91 | 1142 | 6.618 | 19.0 | 6J | 30.7 | -177.0 | 2.15 | 1737 | 10.268 | 50.6 |
| 4E | 34.5 | -26.1 | 1.95 | 1479 | 9.475 | 40.6 | 7A | 38.8 | -210.1 | 1.96 | 1495 | 9.762 | 43.3 |
| 4F | 39.4 | -264.7 | 1.95 | 1573 | 9.663 | 42.2 | 7D | 32.2 | 151.0 | 1.88 | 1663 | 9.172 | 37.1 |
| 4G | 32.1 | -79.6 | 2.02 | 1677 | 9.964 | 45.9 | 7F | 35.9 | -380.6 | 2.09 | 1511 | 9.852 | 45.7 |
| 4H | 43.6 | -335.0 | 2.12 | 1567 | 9.614 | 43.9 | 7G | 31.8 | -183.1 | 2.17 | 1732 | 10.325 | 51.4 |
| 4J | 37.7 | -134.6 | 2.19 | 1775 | 10.133 | 49.7 | 7H | 39.6 | -444.7 | 2.01 | 1592 | 8.956 | 36.8 |
| 5D | 35.7 | 543.0 | 1.63 | 1464 | 8.174 | 27.9 | 7I | 40.1 | -435.1 | 2.20 | 1612 | 9.932 | 47.9 |
| 5F | 31.6 | -491.2 | 2.03 | 1312 | 9.248 | 39.5 | 7J | 33.5 | -249.0 | 2.27 | 1758 | 10.075 | 50.1 |
| 5H | 32.0 | -626.1 | 2.28 | 1424 | 9.956 | 49.0 | CL-20 | 35.8 | 466.5 | 2.07 | 1618 | 9.796 | 46.1 |
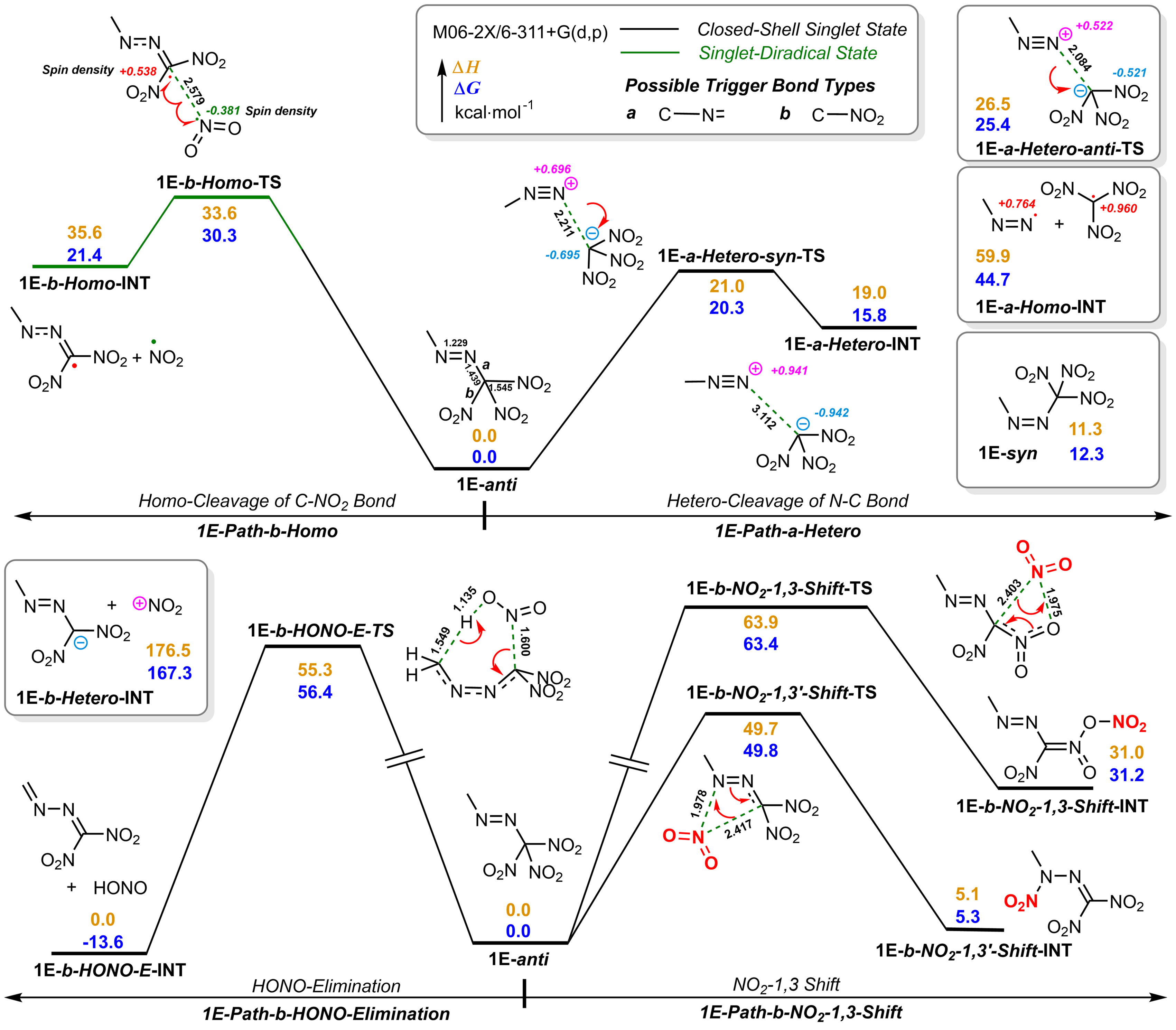
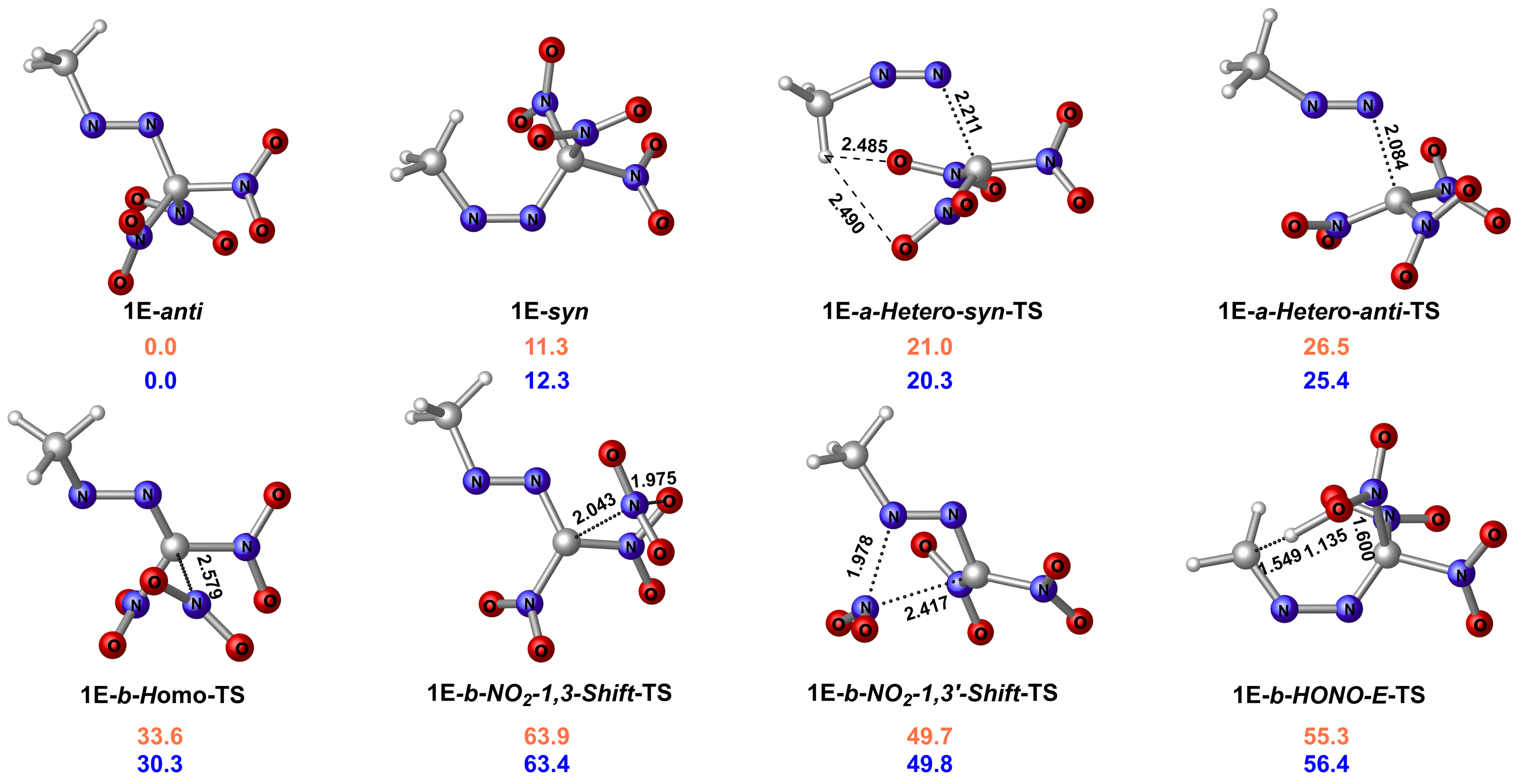
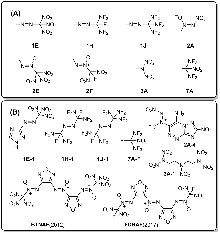
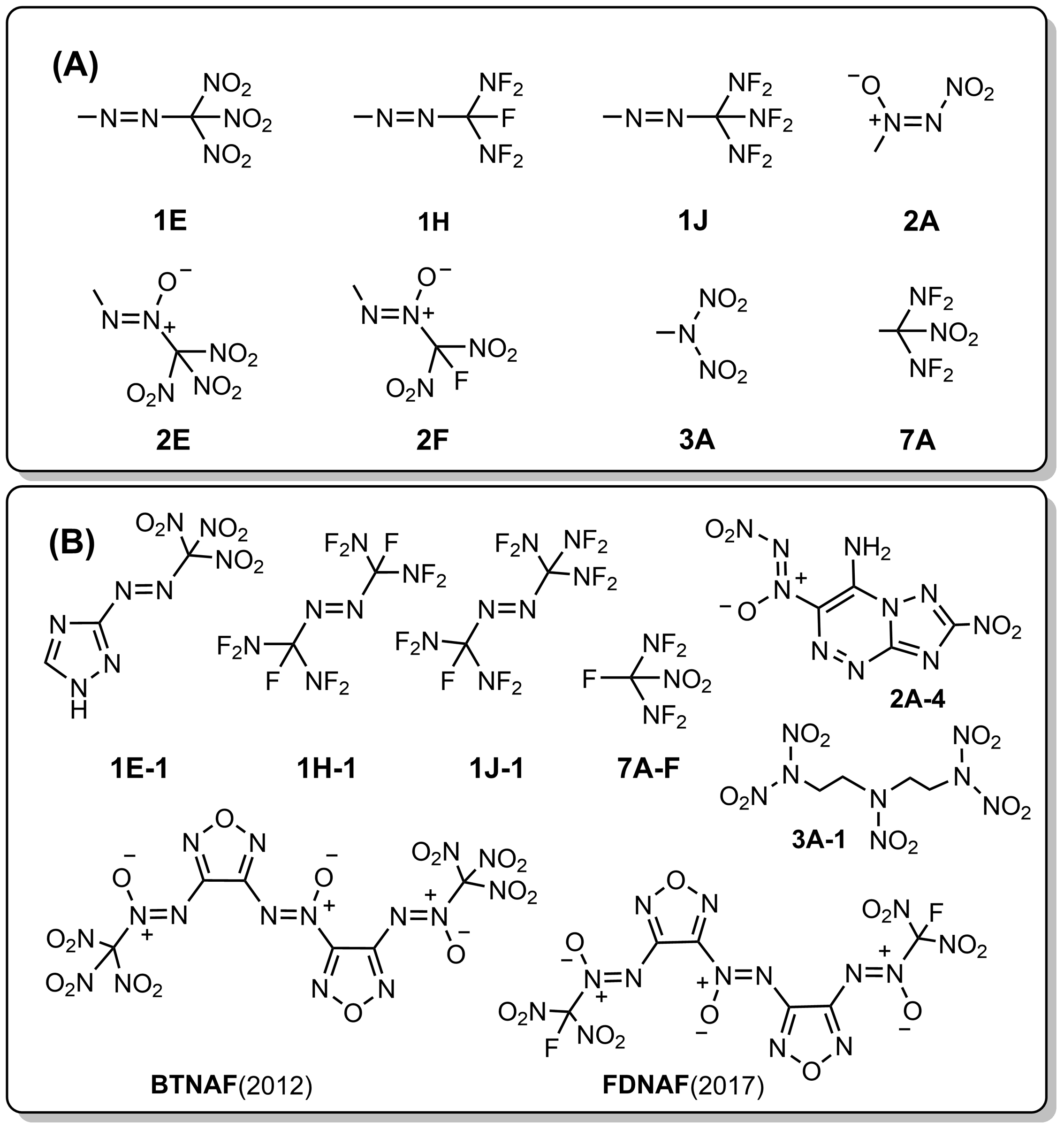
| Group | Compd. | Stability | Ref. | Barrier/ (kcal•mol-1) |
|---|---|---|---|---|
| 1E | 1E-1 | 室温下几天内分解 | [ | 20.3 |
| 1H | 1H-3 | “沸点81 ℃”, “remarkable thermal stability” | [ | 39.2 |
| 1J | 1J-1 | 核磁\色谱观察到很少量样品 | [ | 21.6 |
| 2A | 2A-4 | DSC, Tp=164 ℃ | [ | 33.7 |
| 2E | BTNAF | DSC, Tp=183.6 ℃ | [ | 35.9 |
| 2F | FDNAF | DSC, Tp=233.4 ℃ | [ | 51.1 |
| 3A | 3A-1 | 在熔点分解(71~72 ℃) | [ | 26.7 |
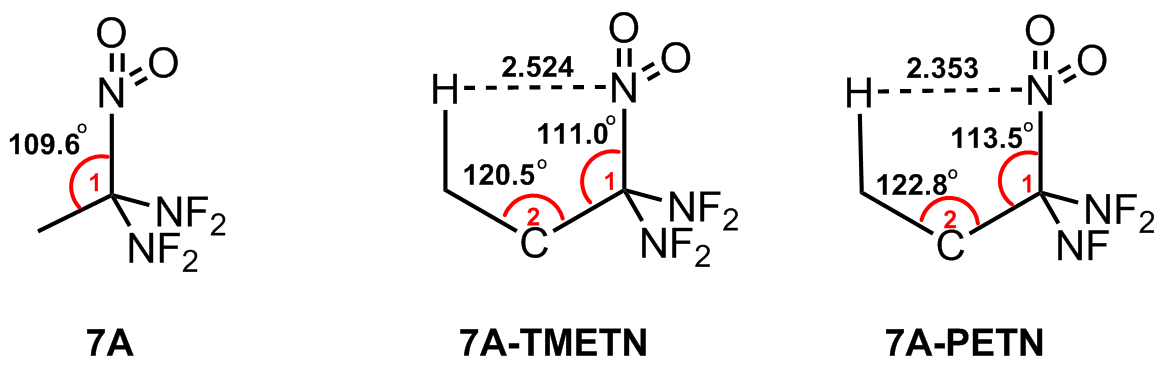
| 7A | 7A-F | 室温数月没有观察到分解 | [ | 38.8 |
| Group | Compd. | Stability | Ref. | Barrier/ (kcal•mol-1) |
|---|---|---|---|---|
| 1E | 1E-1 | 室温下几天内分解 | [ | 20.3 |
| 1H | 1H-3 | “沸点81 ℃”, “remarkable thermal stability” | [ | 39.2 |
| 1J | 1J-1 | 核磁\色谱观察到很少量样品 | [ | 21.6 |
| 2A | 2A-4 | DSC, Tp=164 ℃ | [ | 33.7 |
| 2E | BTNAF | DSC, Tp=183.6 ℃ | [ | 35.9 |
| 2F | FDNAF | DSC, Tp=233.4 ℃ | [ | 51.1 |
| 3A | 3A-1 | 在熔点分解(71~72 ℃) | [ | 26.7 |
| 7A | 7A-F | 室温数月没有观察到分解 | [ | 38.8 |
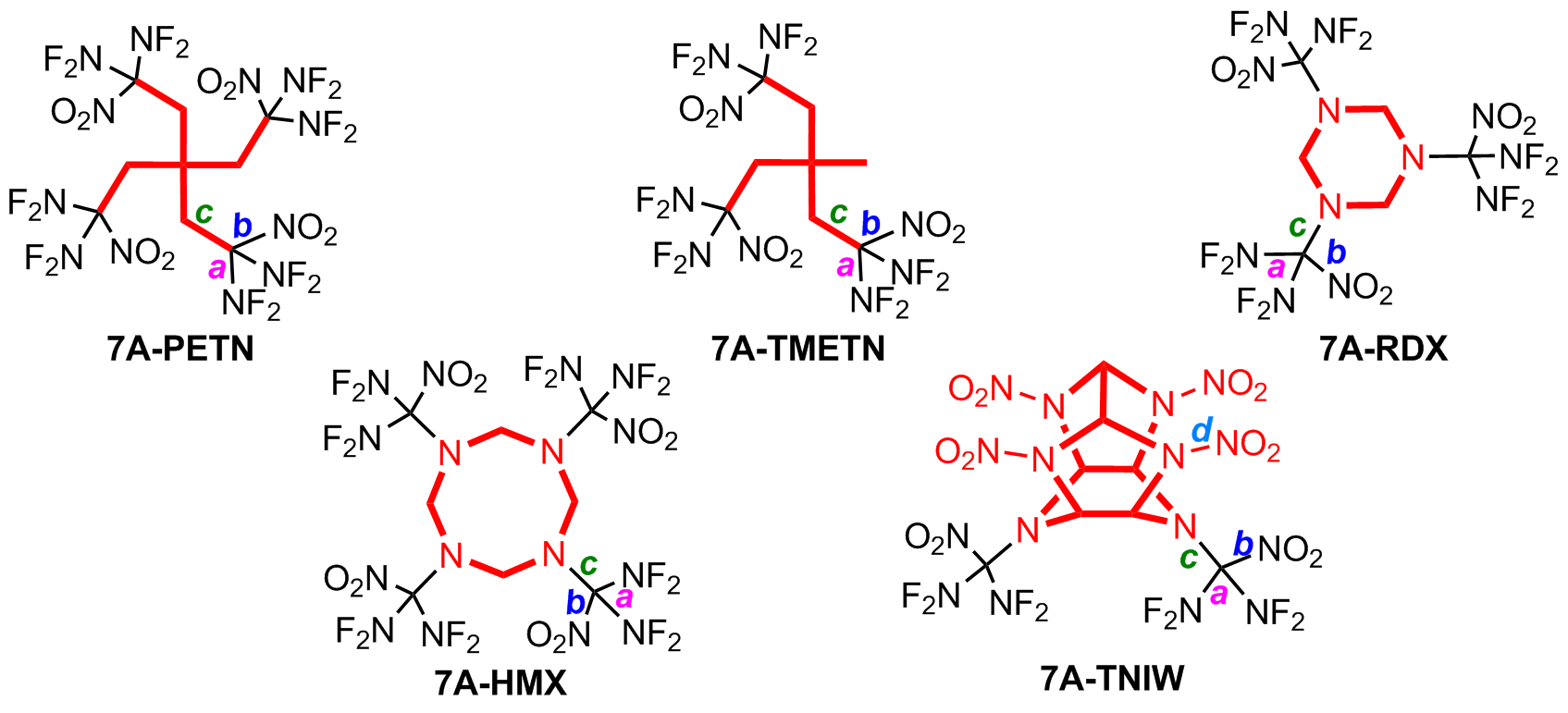
| Compd. | K-J Function | EXPLO5 | ||||
|---|---|---|---|---|---|---|
| D | P | D | P | Isp | ||
| 7A | 9762 | 43.3 | 8427 | 38.8 | 280.2 | |
| 7A-PETN | 10225 | 50.4 | 8679 | 44.1 | 270.3 | |
| 7A-TMETN | 9944 | 46.3 | 8746 | 43.9 | 275.4 | |
| 7A-RDX | 10145 | 49.2 | 8867 | 45.0 | 268.9 | |
| 7A-HMX | 10364 | 52.2 | 9007 | 47.1 | 268.9 | |
| 7A-TNIW | 10297 | 50.8 | 9443 | 48.6 | 278.5 | |
| Cl-20 | 9796 | 46.1 | 9773 | 44.8 | 272.1 | |
| Compd. | K-J Function | EXPLO5 | ||||
|---|---|---|---|---|---|---|
| D | P | D | P | Isp | ||
| 7A | 9762 | 43.3 | 8427 | 38.8 | 280.2 | |
| 7A-PETN | 10225 | 50.4 | 8679 | 44.1 | 270.3 | |
| 7A-TMETN | 9944 | 46.3 | 8746 | 43.9 | 275.4 | |
| 7A-RDX | 10145 | 49.2 | 8867 | 45.0 | 268.9 | |
| 7A-HMX | 10364 | 52.2 | 9007 | 47.1 | 268.9 | |
| 7A-TNIW | 10297 | 50.8 | 9443 | 48.6 | 278.5 | |
| Cl-20 | 9796 | 46.1 | 9773 | 44.8 | 272.1 | |
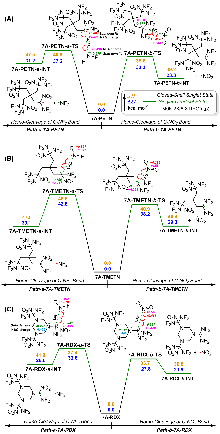
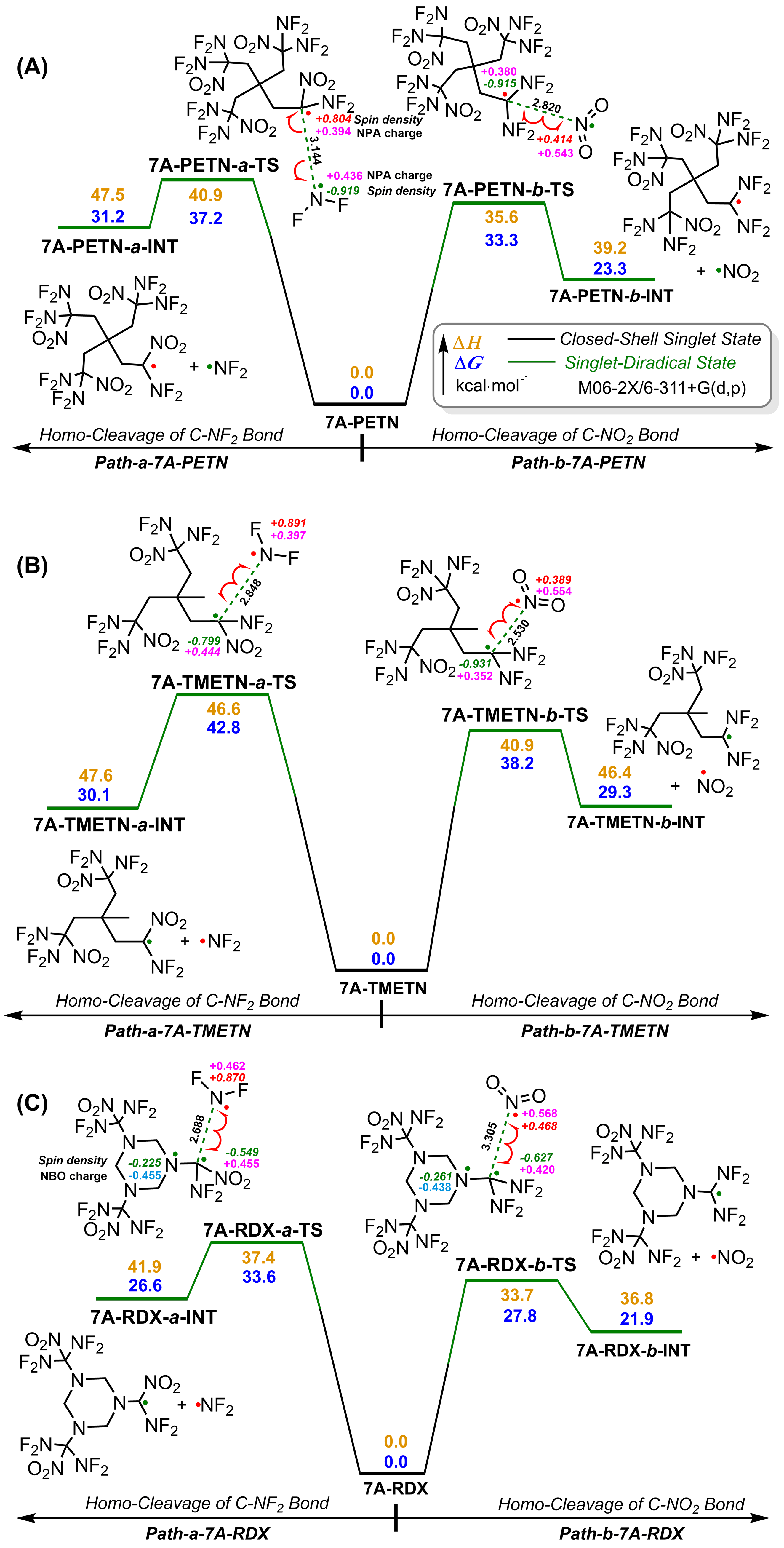
| Compd. | ∆fHSolid | ρ | a/Å | b/Å | c/Å | Barrrier |
|---|---|---|---|---|---|---|
| 7A | -210.1 | 1.96 | 1.480 | 1.541 | — | 38.8 |
| 7A-PETN | -513.0 | 2.17 | 1.491 | 1.536 | 1.539 | 33.3 |
| 7A-TMETN | -497.9 | 2.07 | 1.489 | 1.543 | 1.529 | 38.2 |
| 7A-RDX | -199.1 | 2.14 | 1.523 | 1.563 | 1.382 | 27.8 |
| 7A-HMX | -261.2 | 2.20 | 1.515 | 1.576 | 1.404 | 28.7 |
| 7A-TNIW | 288.1 | 2.14 | 1.507 | 1.585 | 1.403 | 29.9 |
| Compd. | ∆fHSolid | ρ | a/Å | b/Å | c/Å | Barrrier |
|---|---|---|---|---|---|---|
| 7A | -210.1 | 1.96 | 1.480 | 1.541 | — | 38.8 |
| 7A-PETN | -513.0 | 2.17 | 1.491 | 1.536 | 1.539 | 33.3 |
| 7A-TMETN | -497.9 | 2.07 | 1.489 | 1.543 | 1.529 | 38.2 |
| 7A-RDX | -199.1 | 2.14 | 1.523 | 1.563 | 1.382 | 27.8 |
| 7A-HMX | -261.2 | 2.20 | 1.515 | 1.576 | 1.404 | 28.7 |
| 7A-TNIW | 288.1 | 2.14 | 1.507 | 1.585 | 1.403 | 29.9 |
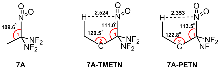
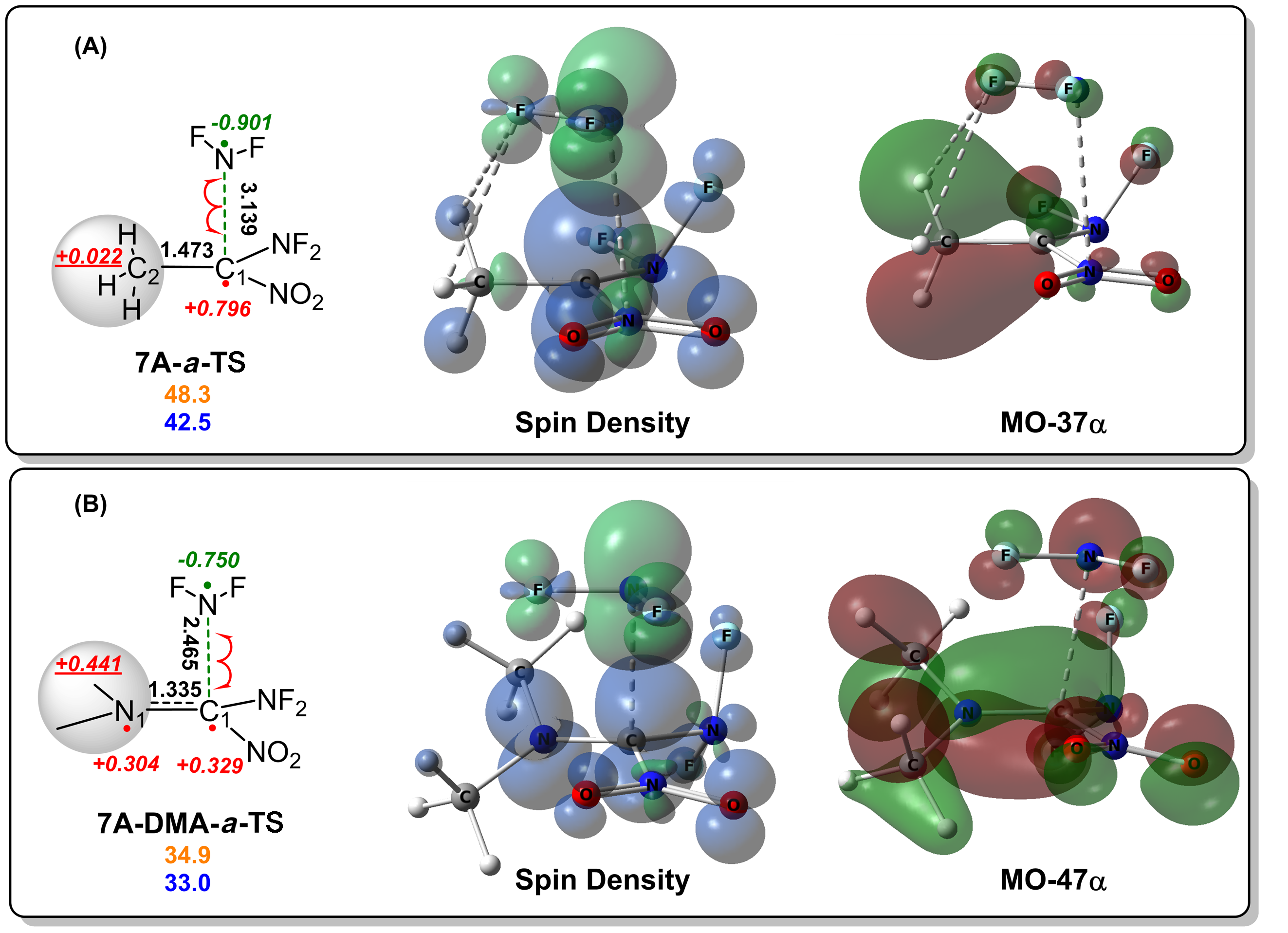
| [1] |
|
| [2] |
|
|
(王文俊, 固体火箭技术, 2003, 26, 42.)
|
|
| [3] |
pmid: 15225926 |
| [4] |
|
|
(黄辉, 王泽山, 黄亨建, 李金山. 火炸药学报, 2005, 28, 9.)
|
|
| [5] |
|
|
(
|
|
| [6] |
doi: 10.1039/C7GC01928A |
| [7] |
|
|
(黄亨建, 黄辉, 中国材料进展, 2018, 37, 889.)
|
|
| [8] |
|
|
(董海山, 含能材料, 2004, A01, 1.)
|
|
| [9] |
|
|
(田均均, 张庆华, 李金山, 含能材料, 2016, 24, 1 )
|
|
| [10] |
doi: 10.1002/prep.v46.6 |
| [11] |
|
| [12] |
doi: 10.1021/ic50066a037 |
| [13] |
doi: 10.1021/ic50108a045 |
| [14] |
doi: 10.1070/MC1996v006n01ABEH000560 |
| [15] |
doi: 10.1007/BF02496149 |
| [16] |
doi: 10.1002/(ISSN)1521-3765 |
| [17] |
doi: 10.5012/bkcs.2013.34.2.686 |
| [18] |
|
| [19] |
doi: 10.1039/C4TA04716H |
| [20] |
|
| [21] |
doi: 10.1002/ejoc.201900314 |
| [22] |
doi: 10.1007/s11172-019-2628-7 |
| [23] |
doi: 10.1002/slct.v5.39 |
| [24] |
doi: 10.1007/s11172-011-0254-0 |
| [25] |
doi: 10.1007/s11172-012-0210-7 |
| [26] |
doi: 10.1007/s11172-012-0049-y |
| [27] |
doi: 10.1007/s11172-012-0050-5 |
| [28] |
doi: 10.1007/s11172-012-0245-9 |
| [29] |
doi: 10.1007/s11172-013-0071-8 |
| [30] |
doi: 10.1007/s11172-015-0832-7 |
| [31] |
doi: 10.1007/s11172-016-1301-7 |
| [32] |
|
|
(张家荣, 毕福强, 王伯周, 张俊林, 翟连杰, 李亚南, 贾思媛, 火炸药学报, 2016, 39, 12.)
doi: 10.14077/j.issn.1007-7812.2016.06.002 |
|
| [33] |
|
|
(毕福强, 王玉, 王伯周, 张家荣, 张俊林, 翟连杰, 李祥志, 含能材料, 2016, 24, 1063.)
|
|
| [34] |
|
| [35] |
|
|
(张家荣, 毕福强, 张俊林, 贾思媛, 火炸药学报, 2017, 40, 45.)
doi: 10.14077/j.issn.1007-7812.2017.05.008 |
|
| [36] |
|
|
(张家荣, 毕福强, 张俊林, 贾思媛, 王伯周, 含能材料, 2021, 29, 798.)
|
|
| [37] |
doi: 10.1007/s11172-015-0824-7 |
| [38] |
|
|
(张家荣, 毕福强, 廉鹏, 张俊林, 王伯周, 有机化学, 2017, 37, 2736.)
doi: 10.6023/cjoc201701014 |
|
| [39] |
|
| [40] |
|
| [41] |
doi: 10.6023/A24010017 |
|
(崔勇康, 成守飞, 凌琳, 李玉学, 吕龙. 化学学报, 2024, 82, 377.)
|
|
| [42] |
|
| [43] |
doi: 10.1016/j.tca.2003.11.019 |
| [44] |
doi: 10.1002/(ISSN)1521-4087 |
| [45] |
|
|
(李卫星, 化学通报, 1988, 6, 19.)
|
|
| [46] |
doi: 10.1021/jacs.9b06576 pmid: 31352783 |
| [47] |
|
| [48] |
doi: 10.1021/ja00260a006 |
| [49] |
doi: 10.1021/jp073117j |
| [50] |
doi: 10.1021/ja00719a006 |
| [51] |
doi: 10.6023/cjoc202206027 |
|
(凌琳, 王健, 李婧, 李玉学, 吕龙, 有机化学, 2023, 43, 285.)
doi: 10.6023/cjoc202206027 |
| [1] | 成守飞, 李婧, 凌琳, 李玉学, 吕龙. 硝化甘油缓慢分解机理的密度泛函理论研究[J]. 化学学报, 2025, 83(8): 833-843. |
| [2] | 马玲玲, 凌琳, 吴亚明, 李玉学, 吕龙. 1,3,5-三乙氧基-2,4,6-三硝基苯的连续化制备反应机理的理论研究[J]. 化学学报, 2025, 83(4): 360-368. |
| [3] | 崔勇康, 成守飞, 凌琳, 李玉学, 吕龙. 二氟氨基二硝甲基芳香杂环含能材料的理论研究[J]. 化学学报, 2024, 82(4): 377-386. |
| [4] | 贾凌轩, 詹泽庞, 贺紫晗, 狄重安, 朱道本. 面向神经电子接口器件的有机材料进展与展望★[J]. 化学学报, 2023, 81(9): 1175-1186. |
| [5] | 李善武, 朱陈宇杰, 罗尹豪, 张亚茹, 滕汉明, 王宗瑞, 甄永刚. 酰胺与酰亚胺类n型有机半导体材料的研究进展[J]. 化学学报, 2022, 80(12): 1600-1617. |
| [6] | 吕敏, 周瑞敏, 吕琨, 魏志祥. 高结晶性小分子给体材料应用于全小分子有机太阳能电池中的研究进展[J]. 化学学报, 2021, 79(3): 284-302. |
| [7] | 母伟花, 马瑶, 方德彩, 王蓉, 张海娜. 1-碘-2-锂-邻碳硼烷与环戊二烯衍生物的类Diels-Alder反应的理论研究[J]. 化学学报, 2018, 76(1): 55-61. |
| [8] | 史清华, 彭谦, 孙少瑞, 帅志刚. 蓝光材料Ir(III)配合物的磷光效率与光谱的振动关联函数研究[J]. 化学学报, 2013, 71(06): 884-891. |
| [9] | 孟祥军, 郭小松, 贾俊芳, 和芹, 王一波. 5水合甘氨酸复合体的结构和性能的理论研究[J]. 化学学报, 2011, 69(01): 25-36. |
| [10] | 李亚娟, 詹晖, 黄可龙, 周运鸿. PEO基锂硫二次聚合物电池[J]. 化学学报, 2010, 68(18): 1850-1854. |
| [11] | 胡建平, 张小轶, 唐典勇, 常珊. HIV-1整合酶与芳香二酮酸类抑制剂相互作用的分子模拟研究[J]. 化学学报, 2009, 67(19): 2177-2183. |
| [12] | 石国升 丁益宏. 双取代铵氧化物(R2HNO)与双取代羟胺(R2NOH)的 相互转换机制的量子化学研究[J]. 化学学报, 2008, 66(22): 2483-2488. |
| [13] | 殷明,舒远杰,雄鹰,罗世凯,龙新平,朱祖良. 硝基咪唑化合物结构与性质的理论研究[J]. 化学学报, 2008, 66(19): 2117-2123. |
| [14] | 周中军 刘慧玲 黄旭日 孙家锺. 预测[Si, C, S]+和[Si, C, S]-体系的稳定异构体[J]. 化学学报, 2008, 66(14): 1637-1640. |
| [15] | 林宪杰, 徐为人, 武剑, 刘成卜. 苯甲醛肟与炔丙醇偶极环加成反应的理论研究[J]. 化学学报, 2007, 65(10): 930-936. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
