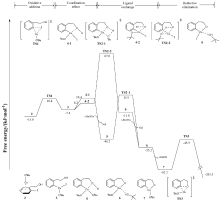
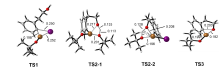
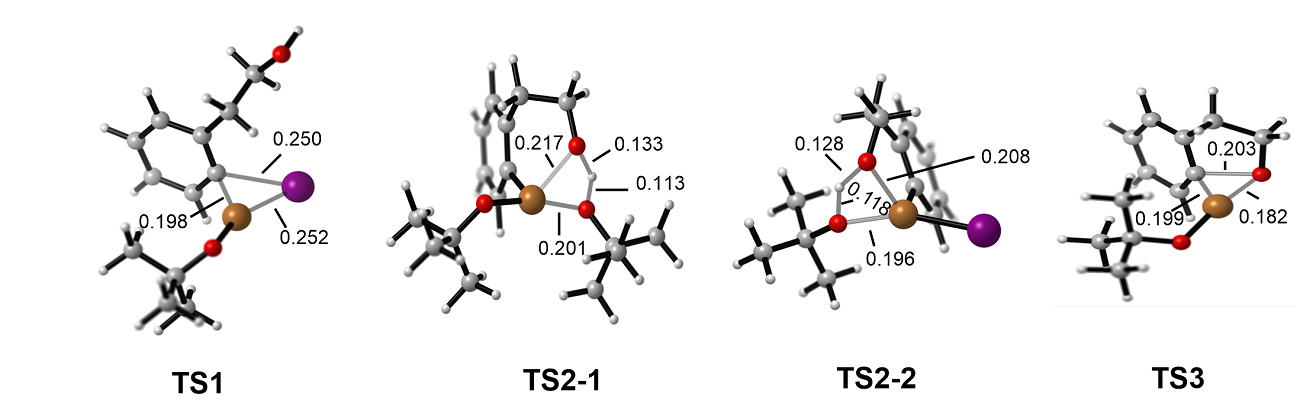
| [1] |
(a) Evano, G.; Wang, J. J.; Nitelet, A. Org. Chem. Front. 2017,2480.
|
|
(b) Ding, H.-W.; Li, J.; Guo, Q.-H.; Xiao, Y. Chinese J. Org. Chem. 2017, 37, 3112. (in Chinese)
doi: 10.6023/cjoc201706006
|
|
(丁怀伟, 李娟, 郭庆辉, 肖琰, 有机化学, 2017, 37, 3112.)
doi: 10.6023/cjoc201706006
|
| [2] |
(a) Liu, Y. J.; Zhang, D. M.; Xiao, S. H.; Qi, Y.; Liu, S. F. Asian J. Org. Chem. 2019, 8, 858.
doi: 10.1002/ajoc.v8.6
|
|
(b) Merritt, J. M.; Andiappan, M.; Pietz, M. A.; Richey, R. N.; Sullivan, K. A.; Kjell, D. P. Org. Process Res. Dev. 2016, 20, 178.
doi: 10.1021/acs.oprd.5b00324
|
|
(c) Monnier, F.; Taillefer, M. Angew. Chem. Int. Ed. 2009, 48, 6954.
doi: 10.1002/anie.v48:38
|
|
(d) Cho, G. Y.; Remy, P.; Jansson, J.; Moessner, C.; Bolm, C. Org. Lett. 2004, 6, 3293.
doi: 10.1021/ol048806h
|
| [3] |
Yang, J.-P.; Zhang, L.; Jin, X.-P.; Gao, H.-Q.; Fang, J.-H.; Li, R.-F.; Fang, Y.-W. Chinese J. Org. Chem. 2013, 33, 1647. (in Chinese)
doi: 10.6023/cjoc201301016
|
|
(杨建平, 张莉, 金小平, 高浩其, 房江华, 李瑞丰, 方烨汶, 有机化学, 2013, 33, 1647.)
doi: 10.6023/cjoc201301016
|
| [4] |
(a) Ouali, A.; Taillefer, M.; Spindler, J. F.; Jutand, A. Organometallics 2007, 26, 65.
doi: 10.1021/om060706n
|
|
(b) Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525.
doi: 10.1039/C3CS60289C
|
| [5] |
Weingarten, H. J. Org. Chem. 1964, 29, 3624.
doi: 10.1021/jo01035a046
|
| [6] |
Paine, A. J. J. Am. Chem. Soc. 1987, 109, 1496.
doi: 10.1021/ja00239a032
|
| [7] |
Marcoux, J. F.; Doye, S.; Buchwald, S. L. J. Am. Chem. Soc. 1997, 119, 10539.
doi: 10.1021/ja971901j
|
| [8] |
Mayoral, J. A.; Rodriguez-Rodriguez, S.; Salvatella, L. Chem. Eur. J. 2008, 14, 9274.
doi: 10.1002/chem.v14:30
|
| [9] |
Tye, J. W.; Weng, Z.; Johns, A. M.; Incarvito, C. D.; Hartwig, J. F. J. Am. Chem. Soc. 2008, 130, 9971.
doi: 10.1021/ja076668w
|
| [10] |
Yu, H. Z.; Jiang, Y. Y.; Fu, Y.; Liu, L. J. Am. Chem. Soc. 2010, 132, 18078.
doi: 10.1021/ja104264v
|
| [11] |
Chen, Z. X.; Jiang, Y. W.; Zhang, L.; Guo, Y. L.; Ma, D. W. J. Am. Chem. Soc. 2019, 141, 3541.
doi: 10.1021/jacs.8b12142
|
| [12] |
Legon, A. C. Angew. Chem, Int. Ed. 1999, 38, 2687.
|
| [13] |
Lefevre, G.; Franc, G.; Adamo, C.; Jutand, A.; Ciofini, I. Organometallics 2012, 31, 914.
doi: 10.1021/om200952v
|
| [14] |
(a) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
pmid: 9944570
|
|
(b) Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Chem. Phys. Lett. 1989, 157, 200.
doi: 10.1016/0009-2614(89)87234-3
pmid: 9944570
|
|
(c) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
doi: 10.1063/1.464913
pmid: 9944570
|
|
(d) Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623.
doi: 10.1021/j100096a001
pmid: 9944570
|
| [15] |
(a) Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257.
doi: 10.1063/1.1677527
|
|
(b) Ditchfield, R.; Hehre, W. J.; Pople, J. A. J. Chem. Phys. 1971, 54, 724.
doi: 10.1063/1.1674902
|
| [16] |
Bergner, A.; Dolg, M.; Kuchle, W.; Stoll, H.; Preuss, H. Mol. Phys. 1993, 80, 1431.
doi: 10.1080/00268979300103121
|
| [17] |
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104.
doi: 10.1063/1.3382344
|
| [18] |
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378.
doi: 10.1021/jp810292n
|
| [19] |
(a) Jover, J. Phys. Chem. Chem. Phys. 2017, 19, 29344.
doi: 10.1039/C7CP05709A
|
|
(b) Jover, J. J. Chem. 2015, 2015, 430358.
|
|
(c) Jover, J.; Spuhler, P.; Zhao, L. G.; McArdle, C.; Maseras, F. Catal. Sci. Technol. 2014, 4, 4200.
doi: 10.1039/C4CY00322E
|
|
(d) Jover, J.; Maseras, F. J. Org. Chem. 2014, 79, 11981.
doi: 10.1021/jo501837p
|
|
(e) Jover, J. ACS Catal. 2014, 4, 4389.
doi: 10.1021/cs500872m
|
| [20] |
(a) Fukui, K. J. Phys. Chem. 1970, 74, 4161.
doi: 10.1021/j100717a029
|
|
(b) Fukui, K. Acc. Chem. Res. 1981, 14, 363.
doi: 10.1021/ar00072a001
|
 ), 蒋华良a,b,*(
), 蒋华良a,b,*(