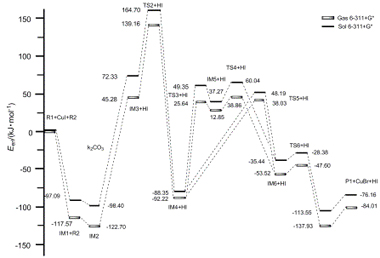
| [1] Sundberg, R. J.; Hegedus, L. S. Angew. Chem., Int. Ed. Engl. 1988, 27, 1113. [2] Gut, I. G.; Wirz, J. Angew Chem., Int. Ed. Engl. 1994, 33, 1153. [3] Taylor, E. C.; Saxton, J. E. The Chemistry of Heterocyclic Compounds, Wiley Interscience, 1983 and supplement, 1994. [4] Miyata, O.; Kimura, Y.; Muroya, K.; Hiramatsu, H.; Naito, T. Tetrahedron Lett. 1999, 40, 3601. [5] Hughes, D. L. Org. Prep. Proc. Int. 1993, 25, 607. [6] Dalpozzo; Renato; Bartoli; Giuseppe Curr. Org. Chem. 2005, 5, 163. [7] Shriner, R. L.; Ashley, W. C.; Welch, E. Org. Synth. Coll. 1955, 3, 725. [8] Rogers, C. V.; Corson, B. B. Org. Synth. Coll. 1963, 5, 884. [9] Takumi, F.; Junya, Y.; Yuki, K.; Ayano, H. J. Org. Chem. 2010, 75, 7010. [10] Hassan, H.; Martine, S.; Frédéric, B.; Cyril, A. J. Org. Chem. 2012, 77(1), 417. [11] Shahzad, S. A.; Vivant, C.; Wirth, T. Org. Lett. 2010, 12, 1364. [12] Schmidt, A. M.; Eilbracht, P. J. Org. Chem. 2005, 70, 5528. [13] Edulji, S. K.; Nguyen, S. T. Organometallics 2003, 22, 3374. [14] Hirotaka, S.; Yusuke, A.; Yoshinori, A.; Yoshiya, F.; Naoto, C. J. Am. Chem. Soc. 2011, 133, 14952. [15] Jian, C.; Xian, H. Org. Lett. 2010, 12, 5048. [16] Jitender, B.; Bariwal, D. S.; Toma, N. Org. Lett. 2010, 12, 2774. [17] Bates, C. G.; Saejueng, P.; Murphy, M. J.; Venkataraman, D. Org. Lett. 2002, 4, 4727. [18] Cacchi, S. Org. Lett. 2003, 5, 3843. [19] Liu, F.; Ma, D, W. J. Org. Chem. 2007, 72, 4844.[20] Zheng, W. R.; Fu, Y.; Wang, H. J.; Guo, Q. X. Chin. J. Org. Chem. 2008, 28, 459 (in Chinese).(郑文锐, 傅尧, 王华静, 郭庆祥, 有机化学, 2008, 28, 459.) [21] Jiang, L. H.; Li, Z. Z.; Chen, C. Y.; Yin, K.; Chen, J. W.; Wang, X. Y.; Huang, Y.; Xiang, J. N. Chin. J. Org. Chem. 2007, 27, 1096 (in Chinese). (蒋历辉, 李治章, 陈超越, 尹凯, 陈金文, 王小勇, 黄勇, 向建南, 有机化学, 2007, 27, 1096.)[22] Nie, H.; Li, Q.; Zhao, K. Q. Chin. J. Org. Chem. 2012, 32, 121 (in Chinese).(聂汉, 李权, 赵可清, 有机化学, 2012, 32, 121.)[23] Liang, X. Q.; Pu, X. M.; Tian, A. M. Chin. J. Org. Chem. 2011, 31, 328 (in Chinese).(梁晓琴, 蒲雪梅, 田安民, 有机化学, 2011, 31, 328.)[24] Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133. [25] Hariharan, P. C.; Pople, J. A. Theor. Chim. Acta 1973, 28, 213. [26] Reed, A. E.; Weinhold, F.; Curtiss, L. A. J. Chem. Phys. 1986, 84, 5687. [27] Barone, V.; Cossi, M. J. Phys. Chem. A 1998, 102,1995. [28] Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, Jr J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision A02, Gaussian, Inc., Wallingford C. T, 2009. |