化学学报 ›› 2011, Vol. 69 ›› Issue (11): 1293-1300. 上一篇 下一篇
研究论文
陈玉红*,1,2,张致龙2,任宝兴2,张材荣2,杜瑞2,王伟超2
Chen Yuhong*,1,2 Zhang Zhilong2 Ren Baoxing2 Zhang Cairong2 Du Rui2 Wang Weichao2
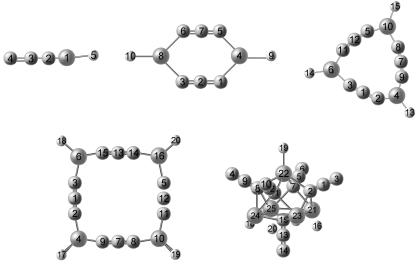
利用密度泛函理论在B3LYP/6-311G*水平上对叠氮化合物(HCaN3)n (n=1~5)团簇各种可能构型进行了几何优化, 预测了各团簇的最稳定结构. 并对最稳定结构的成键特性、电荷分布、振动特性及稳定性进行理论研究. 结果表明, HCaN3团簇最稳定结构为折线型, (HCaN3)n (n=2~4)团簇最稳定结构为叠氮基端位N原子与Ca原子相互链接形成平面环状结构, (HCaN3)5团簇最稳定结构为立体钟形结构. 团簇最稳定结构中金属Ca原子均显示正电性, H原子均显示负电性, 叠氮基中间的N原子显示正电性, 叠氮基两端的N原子显示负电性, 且和Ca原子直接作用的N原子的负电性更强. Ca—N键和Ca—H键为典型的离子键, 叠氮基内N原子之间是共价键. 最稳定结构的IR光谱主要分为3个部分, 其最强振动峰均位于2193~2302 cm-1段, 振动模式为叠氮基中N—N键的反对称伸缩振动. 叠氮基在团簇和晶体中结构不变, 始终以直线型存在, 说明金属叠氮化合物团簇可以很好地模拟其晶体的局域成键和局域电荷转移等特性. 稳定性分析显示, (HCaN3)3团簇相对于其他团簇更稳定.
中图分类号:
