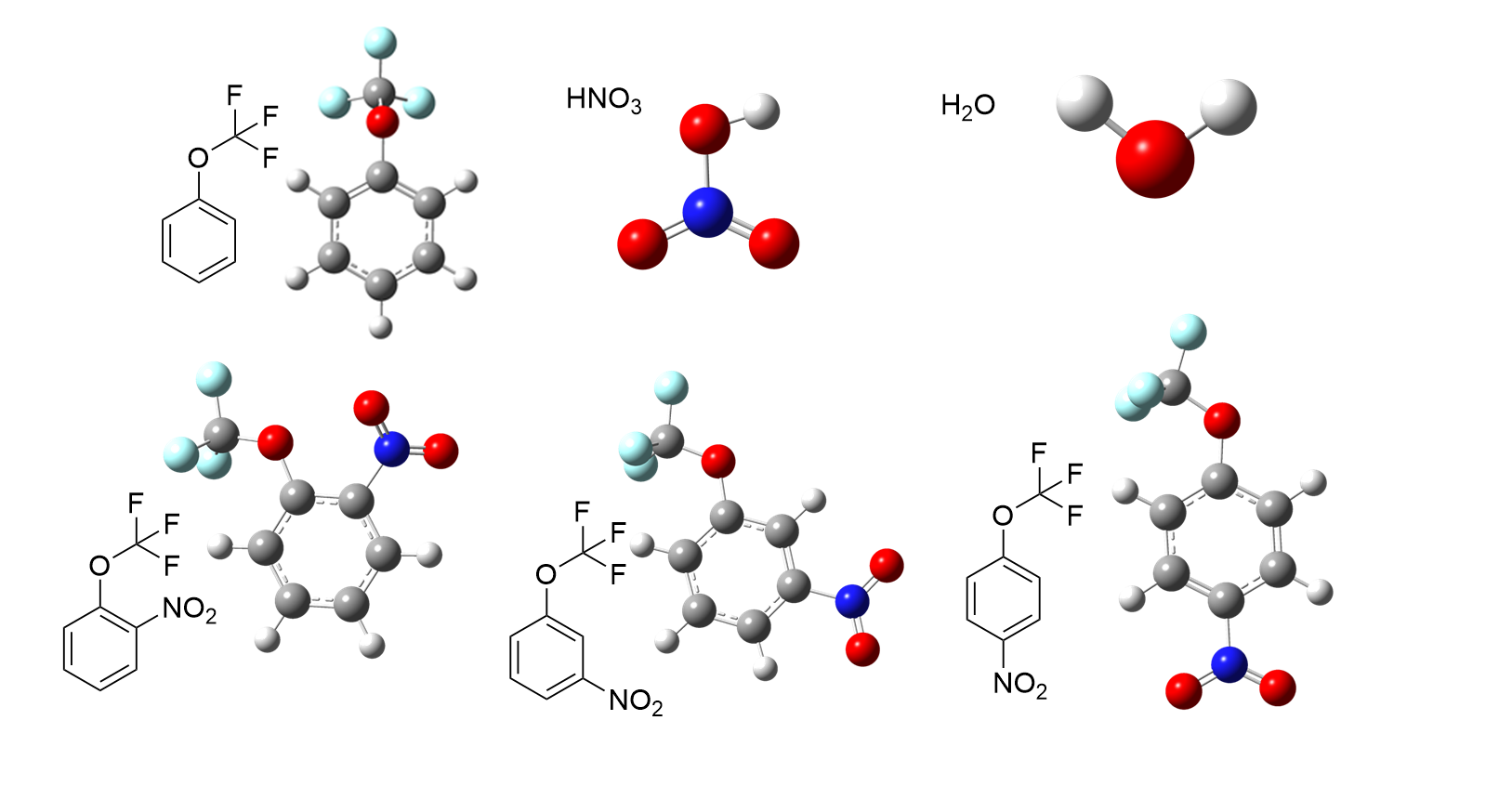
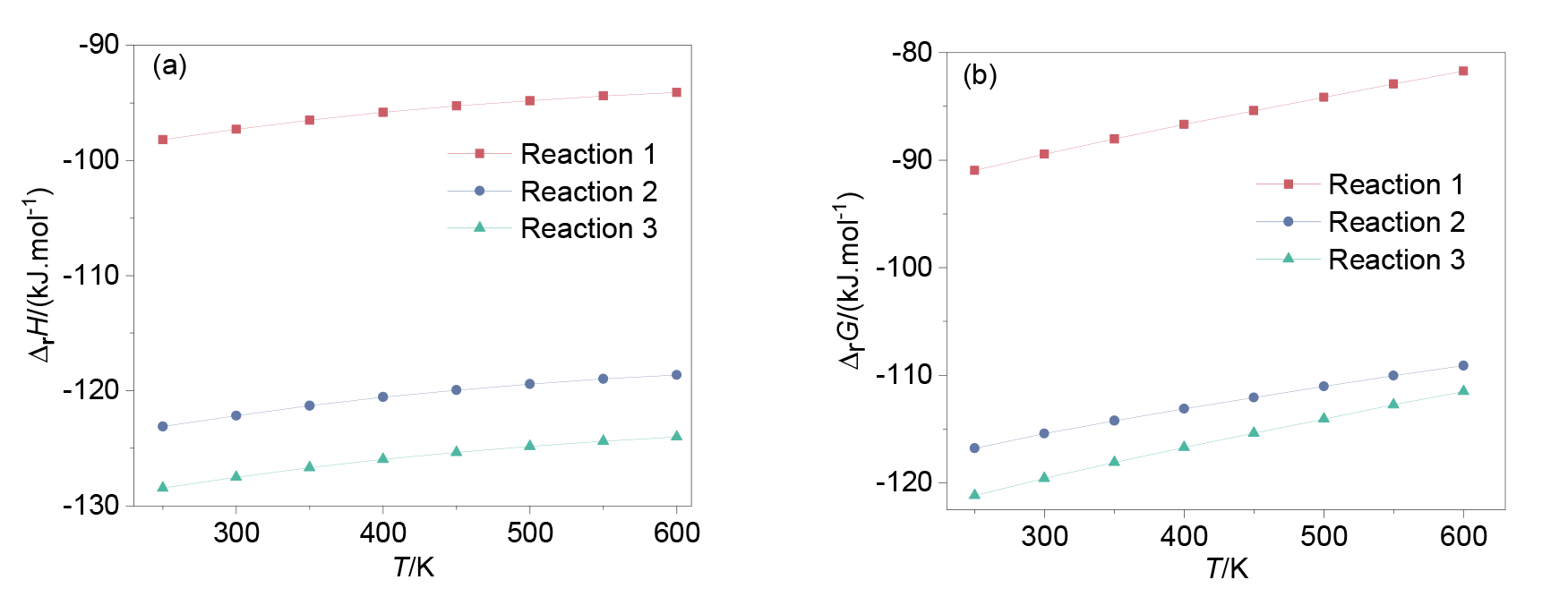
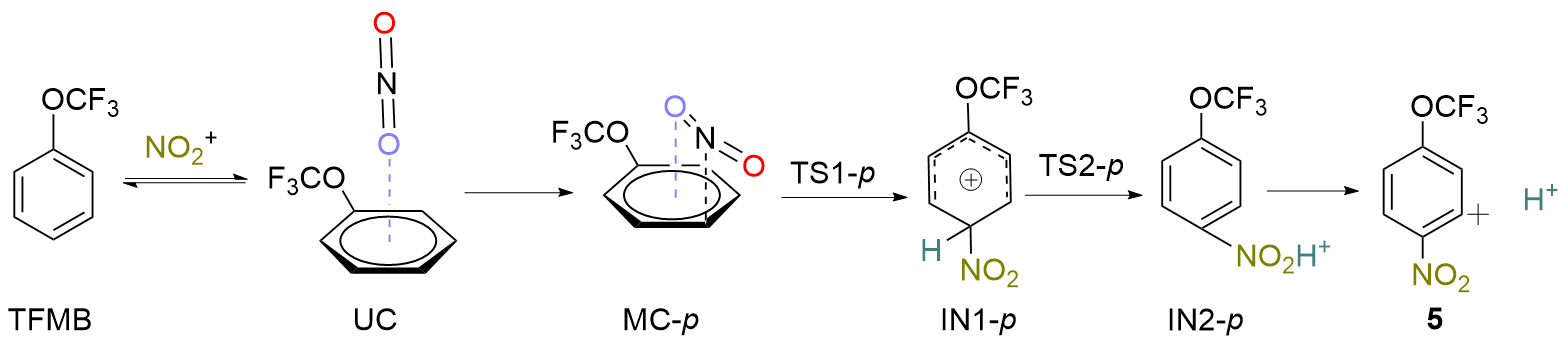
| [1] |
Olah G. A.; Malhotra R.; Narang S. C. Nitration: Methods and Mechanisms, VCH Publishers, New York, 1989, pp. 117-200.
|
| [2] |
Russo D.; Onotri L.; Marotta R.; Andreozzi R.; Somma I. D. Chem. Eng. J. 2017, 307, 1076.
doi: 10.1016/j.cej.2016.09.034
|
| [3] |
Esteves P. M.; Carneiro J. W. M.; Cardoso S. P.; Barbosa A. G. H.; Laali K. K.; Rasul G.; Prakash G. K. S.; Olah G. A. J. Am. Chem. Soc. 2003, 125, 4836.
pmid: 12696903
|
| [4] |
Aitken K. M.; Altken R. A. Sci. Synth. 2009, 40, 1183.
|
| [5] |
Attina M.; Cacace F.; Yanez M. J. Am. Chem. Soc. 1987, 18, 5092.
|
| [6] |
Chen H. H.; Chen L. P.; Wu W. Q.; Guo Z. C. Process Saf. Environ. Prot. 2022, 158, 374.
doi: 10.1016/j.psep.2021.11.036
|
| [7] |
Luo G. S.; Lv Y. C.; Wang K. Micro Chemical Engineering and Technology, Chemical Industry Press, Beijing, 2020, pp. 1-14 (in Chinese).
|
|
(骆广生, 吕阳成, 王凯, 微化工技术, 北京, 化学工业出版社, 2020, pp. 1-14.)
|
| [8] |
Chen L. T.; Xiao H. M.; Xiao J. J.; Gong X. D. J. Phys. Chem. A 2003, 107, 11440.
doi: 10.1021/jp030167p
|
| [9] |
Zhang Q. Z.; Gao R.; Xu F.; Zhou Q.; Ziang G. B.; Wang T.; Chen J. M.; Hu J. T.; Jiang W.; Wang W. X. Environ. Sci. Technol. 2014, 48, 5051.
doi: 10.1021/es500453g
|
| [10] |
Joudah M. K. Res. J. Sci. Technol. 2017, 9, 267.
doi: 10.5958/2349-2988.2017.00048.1
|
| [11] |
Hojczyk K. N.; Feng P. J.; Zhan C. B.; Ngai M. Y. Angew. Chem., Int. Ed. 2014, 53, 14559.
doi: 10.1002/anie.v53.52
|
| [12] |
Wen Z. H.; Yang M.; Zhao S. N.; Zhou F.; Chen G. W. React. Chem. Eng. 2018, 3, 874.
|
| [13] |
Koleva G.; Galabov B.; Hadjieva B.; Schaefer H. F.; Schleyer P. R. Angew. Chem., Int. Ed. 2015, 54, 14123.
doi: 10.1002/anie.v54.47
|
| [14] |
Politzer P.; Jayasuriya K.; Sjoberg P.; Laurence P. R. J. Am. Chem. Soc. 1985, 107, 1174.
doi: 10.1021/ja00291a015
|
| [15] |
Queiroz J. F.; Carneiro J. W.; Sabino A. A.; Sparrapan R.; Eberlin M. N.; Esteves P. M. J. Org. Chem. 2006, 71, 6192.
pmid: 16872205
|
| [16] |
Liljenberg M.; Stenlid J. H.; Brinck T. J. Mol. Model. 2018, 24, 15.
doi: 10.1007/s00894-017-3561-z
|
| [17] |
Cyrille N. N.; Adjieufack A. I.; Maraf M. B.; Charles F. A.; Ibrahim M. N.; Ríos-Gutiérrez M.; Domingo L. R. ChemSelect 2019, 4, 13313.
|
| [18] |
Domingo L. R.; Seif A.; Mazarei E.; Zahedi E.; Ahmadi T. S. Comput. Theor. Chem. 2021, 1199, 113209.
doi: 10.1016/j.comptc.2021.113209
|
| [19] |
Liu S. B. Acta Phys.-Chim. Sin. 2009, 25, 590 (in Chinese).
doi: 10.3866/PKU.WHXB20090332
|
|
(刘述斌, 物理化学学报, 2009, 25, 590.)
|
| [20] |
Geerlings P.; Proft F. D.; Langenaeker W. Chem. Rev. 2003, 103, 1793.
doi: 10.1021/cr990029p
pmid: 12744694
|
| [21] |
Suresh C. H.; Remya G. S.; Anjalikrishna P. K. Wires Comput. Mol. Sci. 2022, 12, 1601.
|
| [22] |
Grimme S. Chem.-Eur. J. 2012, 18, 9955.
doi: 10.1002/chem.v18.32
|
| [23] |
Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A., Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J. Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2015.
|
| [24] |
Liu T. T.; Hu Y. C.; Shen A. Chin. J. Org. Chem. 2023, 43, 622 (in Chinese).
doi: 10.6023/cjoc202206033
|
|
(刘婷婷, 胡宇才, 沈安, 有机化学, 2023, 43, 622.)
doi: 10.6023/cjoc202206033
|
| [25] |
Nieves Q. Y.; Singleton D. A. J. Am. Chem. Soc. 2016, 138, 15167.
pmid: 27794598
|
| [26] |
Shavitt I. Isr. J. Chem. 2013, 33, 357.
doi: 10.1002/ijch.v33.4
|
| [27] |
Yu H. S.; Li S. L.; Truhlar D. G. J. Chem. Phys. 2016, 145, 130901.
doi: 10.1063/1.4963168
|
| [28] |
Hopper D. G. J. Chem. Phys. 1980, 72, 4676.
|
| [29] |
Yao H.; Huang J. L.; Ni Y. Q.; Fu G.; Nie L.; Jiang J. C.; Pan Y. J. Mol. Liq. 2024, 406, 125097.
doi: 10.1016/j.molliq.2024.125097
|
| [30] |
Shi H. C. Eur. J. Chem. 2023, 14, 39.
doi: 10.5155/eurjchem.14.1.39-52.2340
|
| [31] |
Lu T.; Chen Q. X. Comput. Theor. Chem. 2021, 1201, 113249.
|
| [32] |
Alecu I. M.; Zheng J.; Zhao Y.; Truhlar D. G. J. Chem. Theory Comput. 2010, 6, 2872.
doi: 10.1021/ct100326h
pmid: 26616087
|
| [33] |
Lu T.; Chen F. W. J. Comput. Chem. 2012, 33, 580.
doi: 10.1002/jcc.v33.5
|
| [34] |
Lu T. J. Chem. Phys. 2024, 161, 084501.
doi: 10.1063/5.0223879
|
| [35] |
Liu S. B. In Conceptual Density Functional Theory: Towards a New Chemical Reactivity Theory, Vol. 2, Eds.: Lu,T.; ChenQ. X. Wiley-VCHGmbH,Weinheim, 2022, Chapter 31.
|
| [36] |
Fu R.; Lu T.; Chen F. W. Acta Phys.-Chim. Sin. 2014, 30, 628 (in Chinese).
doi: 10.3866/PKU.WHXB201401211
|
|
(付蓉, 卢天, 陈飞武, 物理化学学报, 2014, 30, 628.)
|
| [37] |
Zhang J.; Lu T. Phys. Chem. Chem. Phys. 2021, 23, 20323.
doi: 10.1039/d1cp02805g
pmid: 34486612
|
| [38] |
Domingo L. R.; Ríos-Gutiérrez M.; Pérez P. Molecules 2016, 21, 748.
doi: 10.3390/molecules21060748
|
 ), 赵基钢d,*(
), 赵基钢d,*(