有机化学 ›› 2022, Vol. 42 ›› Issue (5): 1346-1374.DOI: 10.6023/cjoc202110027 上一篇 下一篇
综述与进展
石宇冰a, 白文己a, 母伟花a,*( ), 李江平a, 于嘉玮a, 连冰b
), 李江平a, 于嘉玮a, 连冰b
收稿日期:2021-10-19
修回日期:2021-12-12
发布日期:2022-01-11
通讯作者:
母伟花
基金资助:
Yubing Shia, Wenji Baia, Weihua Mua(), Jiangping Lia, Jiawei Yua, Bing Lianb
Received:2021-10-19
Revised:2021-12-12
Published:2022-01-11
Contact:
Weihua Mu
Supported by:文章分享

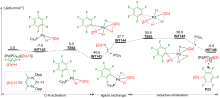
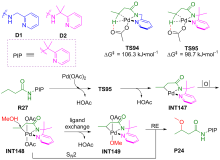
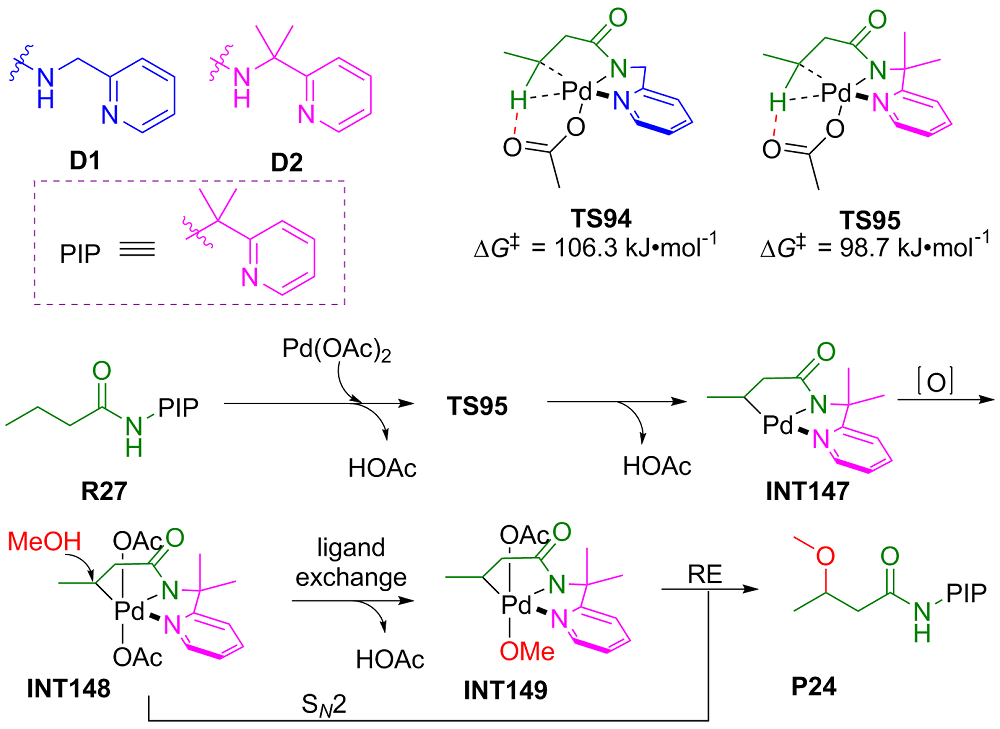
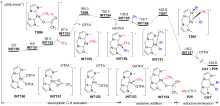
过渡金属催化C—H键官能团化是形成C—X (X=O, N, F, I, ……)键的有效方法, 在传统有机合成、农药、医药以及构筑含C—X杂环的生物活性天然产物基本骨架方面都扮演着重要角色. 钯催化C—H键官能团化形成C—X键的方法具有反应效率高、原子经济性好、环境友好等优点, 是近年来过渡金属催化构建C—X键的研究热点. 采用密度泛函理论(DFT)对钯催化C—H键官能团化形成C—X键的反应开展理论研究, 可获取有关反应路径的诸多信息, 帮助人们从微观层面深入认识此类反应的微观机理和反应选择性调控机制, 为改进钯催化C—H键官能团化形成C—X键的选择性和反应性控制拓展新思路. 对近十年来钯催化C—H键官能团化形成C—X (X=O, N, F, I, ……)键的最新理论研究进展进行了综述, 对上述反应的微观机理和反应选择性进行了深入探讨, 并对该领域的现存问题和发展前景进行了总结与展望.
石宇冰, 白文己, 母伟花, 李江平, 于嘉玮, 连冰. 钯催化C—H键官能团化形成C—X (X=O, N, F, I, ……)键的密度泛函理论研究进展[J]. 有机化学, 2022, 42(5): 1346-1374.
Yubing Shi, Wenji Bai, Weihua Mu, Jiangping Li, Jiawei Yu, Bing Lian. Research Progress on Density Functional Theory Study of Palladium-Catalyzed C—H Functionalization to Form C—X (X=O, N, F, I, …) Bonds[J]. Chinese Journal of Organic Chemistry, 2022, 42(5): 1346-1374.



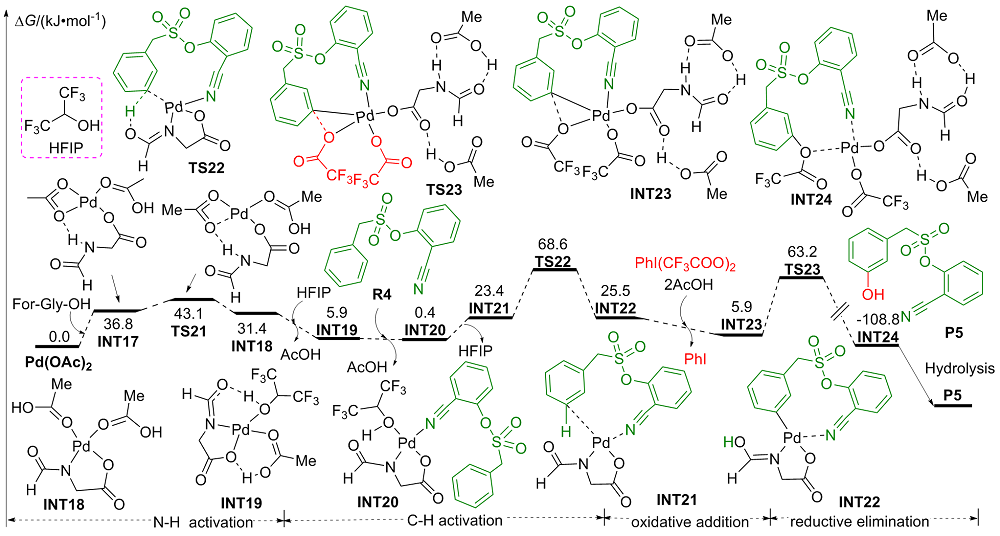


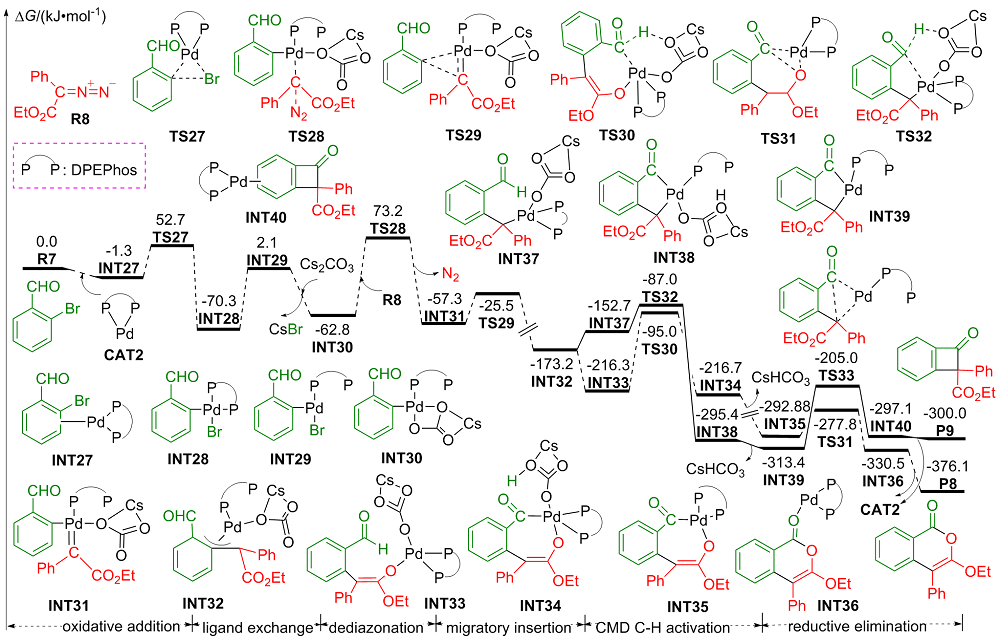


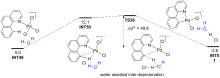


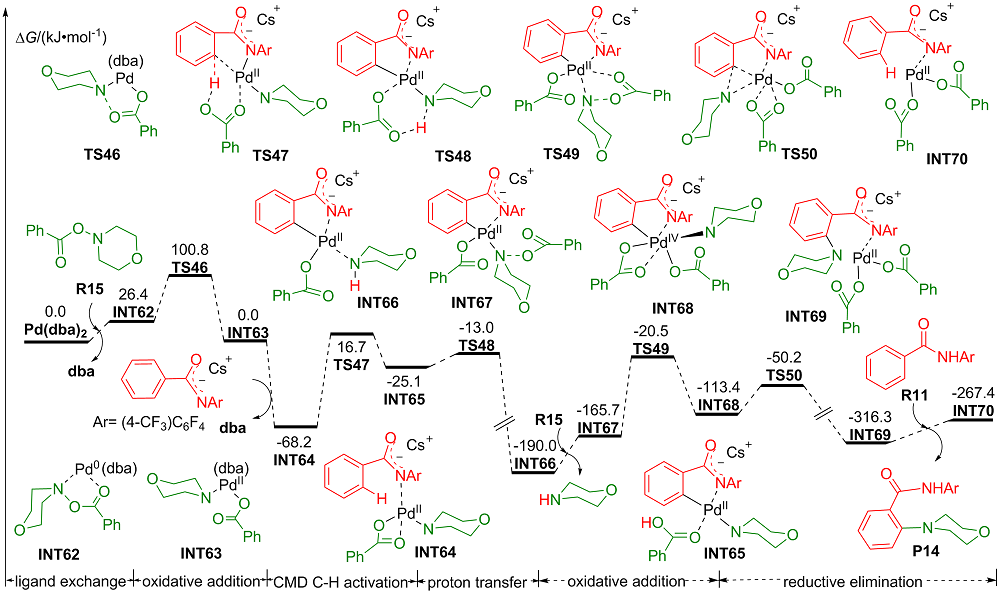
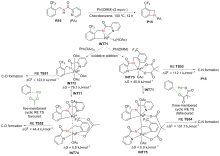


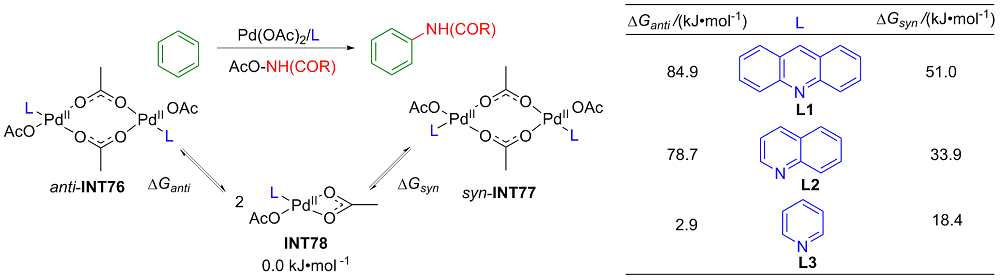
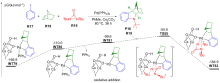










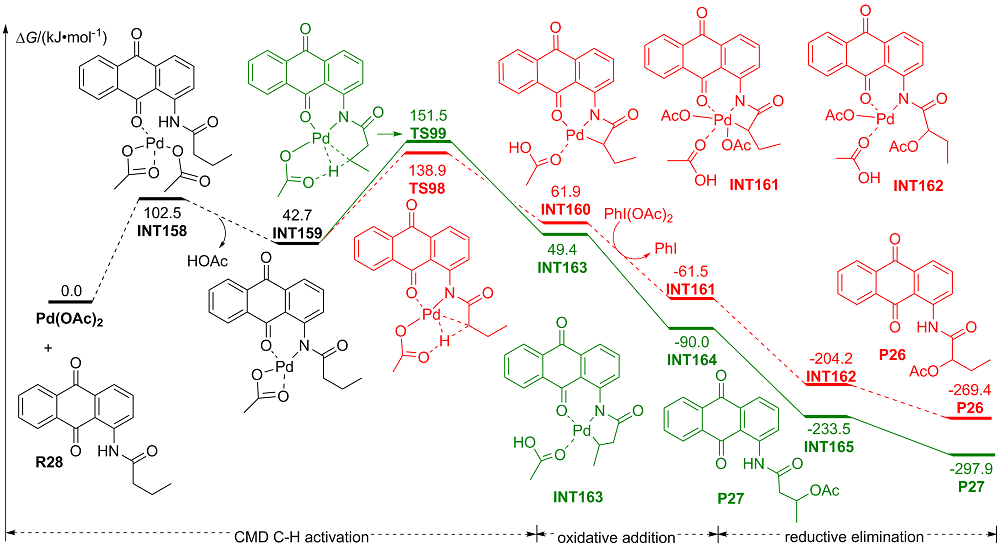
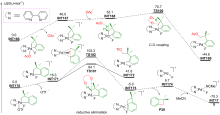

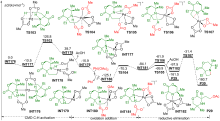



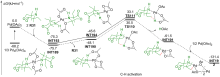
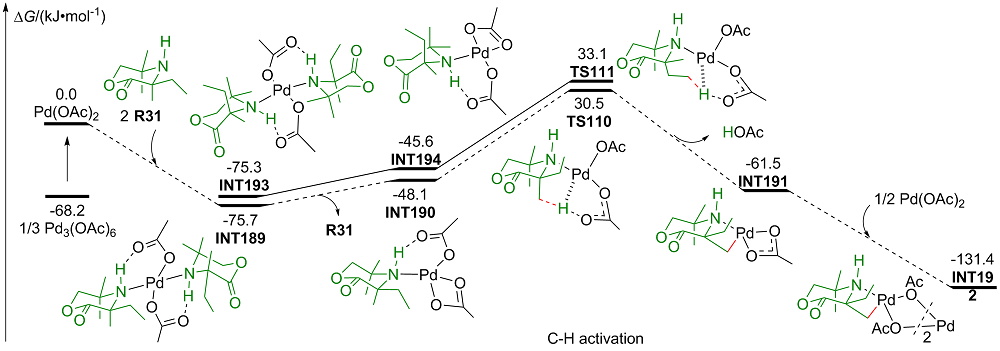




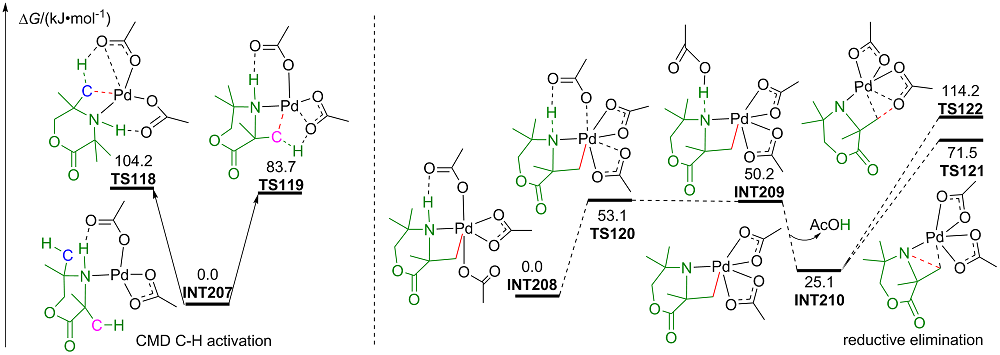
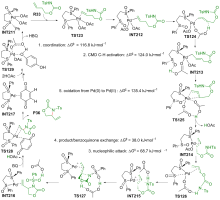

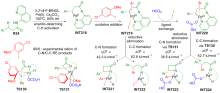

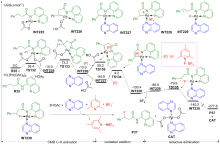

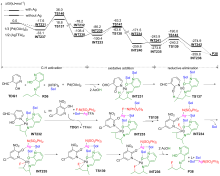

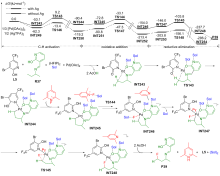
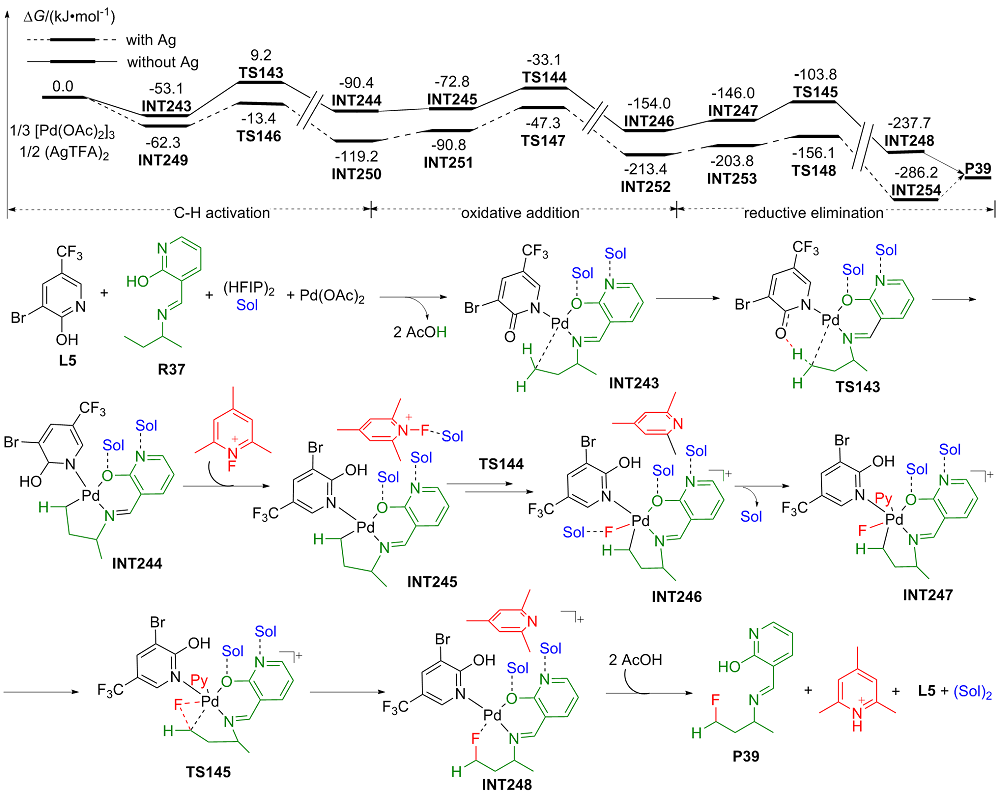

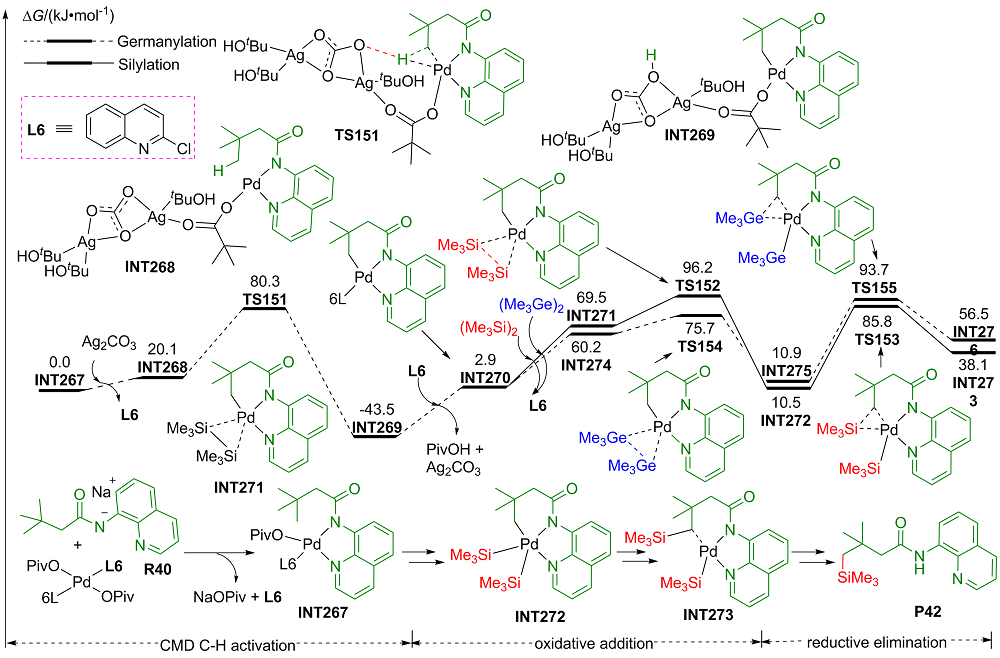
| [1] |
Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094.
doi: 10.1002/anie.200806273 |
| [2] |
Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074.
doi: 10.1021/ar9000058 |
| [3] |
Ackermann, L.; Vicente, R.; Kapdi, A. R. Angew. Chem., Int. Ed. 2009, 48, 9792.
doi: 10.1002/anie.200902996 |
| [4] |
Zhang, S.; Shi, L.; Ding, Y. J. Am. Chem. Soc. 2011, 133, 20218.
doi: 10.1021/ja205294y |
| [5] |
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147.
doi: 10.1021/cr900184e |
| [6] |
Zeni, G.; Larock, R. C. Chem. Rev. 2004, 104, 2285.
doi: 10.1021/cr020085h |
| [7] |
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395.
doi: 10.1021/cr050041j |
| [8] |
Wu, M.-J.; Chu, J.-H. J. Chin. Chem. Soc. 2020, 67, 399.
doi: 10.1002/jccs.201900471 |
| [9] |
Liao, G.; Zhang, T.; Lin, Z.-K.; Shi, B.-F. Angew. Chem., Int. Ed. 2020, 59, 19773.
doi: 10.1002/anie.202008437 |
| [10] |
Gramage-Doria, R. Chem.-Eur. J. 2020, 26, 9688.
doi: 10.1002/chem.202000672 |
| [11] |
Liao, K.; Negretti, S.; Musaev, D. G.; Bacsa, J.; Davies, H. M. L. Nature 2016, 533, 230.
doi: 10.1038/nature17651 |
| [12] |
Kumar, P.; Nagtilak, P. J.; Kapur, M. New J. Chem. 2021, 45, 13692.
doi: 10.1039/D1NJ01696B |
| [13] |
Das, R.; Kapur, M. J. Org. Chem. 2017, 82, 1114.
doi: 10.1021/acs.joc.6b02731 |
| [14] |
Youn, S. W.; Cho, C.-G. Org. Biomol. Chem. 2021, 19, 5028.
doi: 10.1039/d1ob00506e pmid: 34027964 |
| [15] |
Luo, H.-H.; Pei, N.; Zhang, J. Chin. J. Org. Chem. 2021, 41, 2990. (in Chinese)
doi: 10.6023/cjoc202103013 |
|
(罗欢欢, 裴娜, 张敬, 有机化学, 2021, 41, 2990.)
doi: 10.6023/cjoc202103013 |
|
| [16] |
Zou, X.-L.; Xu, X.-M. Chin. J. Org. Chem. 2021, 41, 2610. (in Chinese)
doi: 10.6023/cjoc202103020 |
|
(邹晓亮, 徐森苗, 有机化学, 2021, 41, 2610.)
doi: 10.6023/cjoc202103020 |
|
| [17] |
Marchese, A. D.; Adrianov, T.; Lautens, M. Angew. Chem., Int. Ed. 2021, 60, 16750.
doi: 10.1002/anie.202101324 |
| [18] |
Vijaykumar, M.; Punji, B. Synthesis 2021, 53, 2935.
doi: 10.1055/a-1481-2584 |
| [19] |
Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem., Int. Ed. 2012, 51, 8960.
doi: 10.1002/anie.201201666 |
| [20] |
Newhouse, T.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 3362.
doi: 10.1002/anie.201006368 |
| [21] |
Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. Eur. J. 2010, 16, 2654.
doi: 10.1002/chem.200902374 |
| [22] |
Chen, D. Y. K.; Youn, S. W. Chem.-Eur. J. 2012, 18, 9452.
doi: 10.1002/chem.201201329 |
| [23] |
Roughley, S. D.; Jordan, A. M. J. Med. Chem. 2011, 54, 3451.
doi: 10.1021/jm200187y pmid: 21504168 |
| [24] |
Chao, J.; Li, H.; Cheng, K. W.; Yu, M. S.; Chang, R. C.; Wang, M. J. Nutr. Biochem. 2010, 21, 482.
doi: 10.1016/j.jnutbio.2009.02.004 |
| [25] |
Chen, T.-B.; Zhang, M. Chin. J. Org. Chem. 2015, 35, 813. (in Chinese)
doi: 10.6023/cjoc201409022 |
|
(陈天保, 章明, 有机化学, 2015, 35, 813.)
doi: 10.6023/cjoc201409022 |
|
| [26] |
Zhao, k.; Yang, L.; Liu, J.-H.; Xia, C.-G. Chin. J. Org. Chem. 2018, 38, 2833. (in Chinese)
doi: 10.6023/cjoc201805028 |
|
(赵康, 杨磊, 刘建华, 夏春谷, 有机化学, 2018, 38, 2833.)
doi: 10.6023/cjoc201805028 |
|
| [27] |
Timsina, Y. N.; Gupton, B. F.; Ellis, K. C. ACS Catal. 2018, 8, 5732.
doi: 10.1021/acscatal.8b01168 |
| [28] |
Zhang, M.; Wang, Q.; Peng, Y.; Chen, Z.; Wan, C.; Chen, J.; Zhao, Y.; Zhang, R.; Zhang, A. Q. Chem. Commun. 2019, 55, 13048.
doi: 10.1039/C9CC06609H |
| [29] |
Petrone, D. A.; Ye, J.; Lautens, M. Chem. Rev. 2016, 116, 8003.
doi: 10.1021/acs.chemrev.6b00089 pmid: 27341176 |
| [30] |
Das, R.; Kapur, M. Asian J. Org. Chem. 2018, 7, 1524.
doi: 10.1002/ajoc.201800142 |
| [31] |
Ma, X.; Mo, Q.; Chang, J.; Xie, K. Synth. Commun. 2018, 48, 1403.
doi: 10.1080/00397911.2018.1455872 |
| [32] |
Mboyi, C. D.; Testa, C.; Reeb, S.; Genc, S.; Cattey, H.; Fleurat-Lessard, P.; Roger, J.; Hierso, J.-C. ACS Catal. 2017, 7, 8493.
doi: 10.1021/acscatal.7b03186 |
| [33] |
Xu, J.; Wei, Z.; Li, J.-R. Chin. J. Org. Chem. 2012, 32, 1208. (in Chinese)
doi: 10.6023/cjoc1108122 |
|
(徐娟, 魏真, 李加荣, 有机化学, 2012, 32, 1208.)
doi: 10.6023/cjoc1108122 |
|
| [34] |
Zhou, B.; Lu, A.; Zhang, Y. Synlett 2019, 30, 685.
doi: 10.1055/s-0037-1610339 |
| [35] |
Borpatra, P. J.; Deka, B.; Deb, M. L.; Baruah, P. K. Org. Chem. Front. 2019, 6, 3445.
doi: 10.1039/c9qo00863b |
| [36] |
Boller, T. M.; Murphy, J. M.; Hapke, M.; Ishiyama, T.; Miyaura, N.; Hartwig, J. F. J. Am. Chem. Soc. 2005, 127, 14263.
doi: 10.1021/ja053433g |
| [37] |
Plata, R. E.; Singleton, D. A. J. Am. Chem. Soc. 2015, 137, 3811.
doi: 10.1021/ja5111392 pmid: 25714789 |
| [38] |
Bonney, K. J.; Schoenebeck, F. Chem. Soc. Rev. 2014, 43, 6609.
doi: 10.1039/c4cs00061g pmid: 24759955 |
| [39] |
Houk, K. N. Chem. Soc. Rev. 2014, 43, 4905.
doi: 10.1039/c4cs90049a pmid: 24916988 |
| [40] |
Cheng, G.-J.; Zhang, X.; Chung, L. W.; Xu, L.; Wu, Y.-D. J. Am. Chem. Soc. 2015, 137, 1706.
doi: 10.1021/ja5112749 |
| [41] |
Zhang, X.; Chung, L. W.; Wu, Y.-D. Acc. Chem. Res. 2016, 49, 1302.
doi: 10.1021/acs.accounts.6b00093 |
| [42] |
Jiang, Y.-Y.; Man, X.; Bi, S. Sci. China Chem. 2016, 59, 1448.
doi: 10.1007/s11426-016-0330-3 |
| [43] |
Zhang, K.-R.; Wang, Y.-Y.; Zhu, H.-D.; Peng, Q. Chin. J. Org. Chem. 2021, 41, 3995. (in Chinese)
doi: 10.6023/cjoc202102036 |
|
(张凯瑞, 王亚亚, 朱宏丹, 彭谦, 有机化学, 2021, 41, 3995.)
|
|
| [44] |
Balcells, D.; Clot, E.; Eisenstein, O. Chem. Rev. 2010, 110, 749.
doi: 10.1021/cr900315k pmid: 20067255 |
| [45] |
Musaev, D. G.; Figg, T. M.; Kaledin, A. L. Chem. Soc. Rev. 2014, 43, 5009.
doi: 10.1039/C3CS60447K |
| [46] |
Sperger, T.; Sanhueza, I. A.; Kalvet, I.; Schoenebeck, F. Chem. Rev. 2015, 115, 9532.
doi: 10.1021/acs.chemrev.5b00163 |
| [47] |
Davies, D. L.; Macgregor, S. A.; McMullin, C. L. Chem. Rev. 2017, 117, 8649.
doi: 10.1021/acs.chemrev.6b00839 pmid: 28530807 |
| [48] |
Yang, Y.-F.; She, Y. Int J Quantum Chem. 2018, 118, e25723.
doi: 10.1002/qua.25723 |
| [49] |
Xie, H.; Fan, T.; Lei, Q.; Fang, W. Sci. China Chem. 2016, 59, 1432.
doi: 10.1007/s11426-016-0018-2 |
| [50] |
Yang, B.; Schouten, A.; Ess, D. H. J. Am. Chem. Soc. 2021, 143, 8367.
doi: 10.1021/jacs.1c01709 |
| [51] |
Gygi, D.; Gonzalez, M. I.; Hwang, S. J.; Xia, K. T.; Qin, Y.; Johnson, E. J.; Gygi, F.; Chen, Y.-S.; Nocera, D. G. J. Am. Chem. Soc. 2021, 143, 6060.
doi: 10.1021/jacs.1c02630 |
| [52] |
Esteruelas, M. A.; Martínez, A.; Oliván, M.; Oñate, E. J. Am. Chem. Soc. 2020, 142, 19119.
doi: 10.1021/jacs.0c07578 pmid: 33125215 |
| [53] |
Bera, M.; Maji, A.; Sahoo, S. K.; Maiti, D. Angew. Chem., Int. Ed. 2015, 54, 8515.
doi: 10.1002/anie.201503112 |
| [54] |
He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J.-Q. Chem. Rev. 2017, 117, 8754.
doi: 10.1021/acs.chemrev.6b00622 |
| [55] |
Salazar, C. A.; Gair, J. J.; Flesch, K. N.; Guzei, I. A.; Lewis, J. C.; Stahl, S. S. Angew. Chem., Int. Ed. 2020, 59, 10873.
doi: 10.1002/anie.202002484 |
| [56] |
Bai, Z.; Cai, C.; Sheng, W.; Ren, Y.; Wang, H. Angew. Chem., Int. Ed. 2020, 59, 14686.
doi: 10.1002/anie.202007226 |
| [57] |
Ezawa, T.; Sohtome, Y.; Hashizume, D.; Adachi, M.; Akakabe, M.; Koshino, H.; Sodeoka, M. J. Am. Chem. Soc. 2021, 143, 9094.
doi: 10.1021/jacs.1c02833 |
| [58] |
Oliveira, J. C. A.; Dhawa, U.; Ackermann, L. ACS Catal. 2021, 11, 1505.
doi: 10.1021/acscatal.0c04205 |
| [59] |
Sahharova, L. T.; Gordeev, E. G.; Eremin, D. B.; Ananikov, V. P. ACS Catal. 2020, 10, 9872.
doi: 10.1021/acscatal.0c02053 |
| [60] |
Kang, K.; Huang, L.; Weix, D. J. J. Am. Chem. Soc. 2020, 142, 10634.
doi: 10.1021/jacs.0c04670 pmid: 32486635 |
| [61] |
Yoshino, T.; Matsunaga, S. ACS Catal. 2021, 11, 6455.
doi: 10.1021/acscatal.1c01351 |
| [62] |
Zhu, M.-H.; Zhang, X.-W.; Usman, M.; Cong, H.; Liu, W.-B. ACS Catal. 2021, 11, 5703.
doi: 10.1021/acscatal.1c00975 |
| [63] |
Kato, Y.; Lin, L.; Kojima, M.; Yoshino, T.; Matsunaga, S. ACS Catal. 2021, 11, 4271.
doi: 10.1021/acscatal.1c00765 |
| [64] |
Liu, J.-R.; Duan, Y.-Q.; Zhang, S.-Q.; Zhu, L.-J.; Jiang, Y.-Y.; Bi, S.; Hong, X. Org. Lett. 2019, 21, 2360.
doi: 10.1021/acs.orglett.9b00633 pmid: 30892899 |
| [65] |
Veerakumar, P.; Thanasekaran, P.; Lu, K.-L.; Lin, K.-C.; Rajagopal, S. ACS Sustainable Chem. Eng. 2017, 5, 8475.
doi: 10.1021/acssuschemeng.7b00922 |
| [66] |
Rao, D. Y.; Anoop, A. J. Phys. Chem. C 2020, 124, 582.
doi: 10.1021/acs.jpcc.9b09316 |
| [67] |
Wang, Z.; Fu, Y.; Zhang, Q.; Liu, H.; Wang, J. J. Org. Chem. 2020, 85, 7683.
doi: 10.1021/acs.joc.0c00115 |
| [68] |
Favier, L.; Pla, D.; Gómez, M. Chem. Rev. 2020, 120, 1146.
doi: 10.1021/acs.chemrev.9b00204 |
| [69] |
Anand, M.; Sunoj, R. B. Org. Lett. 2011, 13, 4802.
doi: 10.1021/ol201830r |
| [70] |
Anand, M.; Sunoj, R. B. Organometallics 2012, 31, 6466.
doi: 10.1021/om300681e |
| [71] |
Lian, B.; Zhang, L.; Chass, G. A.; Fang, D.-C. J. Org. Chem. 2013, 78, 8376.
doi: 10.1021/jo4010712 |
| [72] |
Sun, Y.-H.; Sun, T.-Y.; Wu, Y.-D.; Zhang, X.; Rao, Y. Chem. Sci. 2016, 7, 2229.
doi: 10.1039/C5SC03905C |
| [73] |
Maji, A.; Bhaskararao, B.; Singha, S.; Sunoj, R. B.; Maiti, D. Chem. Sci. 2016, 7, 3147.
doi: 10.1039/C5SC04060D |
| [74] |
Simmons, E. M.; Hartwig, J. F. Angew. Chem., Int. Ed. 2012, 51, 3066.
doi: 10.1002/anie.201107334 |
| [75] |
Yang, Z.-W.; Zhang, Q.; Jiang, Y.-Y.; Li, L.; Xiao, B.; Fu, Y. Chem. Commun. 2016, 52, 6709.
doi: 10.1039/C6CC01732K |
| [76] |
Yu, Y.; Lu, Q.; Chen, G.; Li, C.; Huang, X. Angew. Chem., Int. Ed. 2018, 57, 319.
doi: 10.1002/anie.201710317 |
| [77] |
Jiang, J.; Yuan, D.; Ma, C.; Song, W.; Lin, Y.; Hu, L.; Zhang, Y. Org. Lett. 2021, 23, 279.
doi: 10.1021/acs.orglett.0c03701 pmid: 33352055 |
| [78] |
Ke, Z.; Cundari, T. R. Organometallics 2010, 29, 821.
doi: 10.1021/om900895t |
| [79] |
Yoo, E. J.; Ma, S.; Mei, T.-S.; Chan, K. S. L.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 7652.
doi: 10.1021/ja202563w |
| [80] |
Anand, M.; Sunoj, R. B.; Schaefer, H. F. J. Am. Chem. Soc. 2014, 136, 5535.
doi: 10.1021/ja412770h |
| [81] |
Anand, M.; Sunoj, R. B.; Schaefer, H. F. ACS Catal. 2016, 6, 696.
doi: 10.1021/acscatal.5b02639 |
| [82] |
Zhou, Y.; Bao, X. Org. Lett. 2016, 18, 4506.
doi: 10.1021/acs.orglett.6b02093 |
| [83] |
He, G.; Lu, G.; Guo, Z.; Liu, P.; Chen, G. Nature Chem. 2016, 8, 1131.
doi: 10.1038/nchem.2585 |
| [84] |
Gary, J. B.; Sanford, M. S. Organometallics 2011, 30, 6143.
doi: 10.1021/om200677y |
| [85] |
Bandara, H. M. D.; Jin, D.; Mantell, M. A.; Field, K. D.; Wang, A.; Narayanan, R. P.; Deskins, N. A.; Emmert, M. H. Catal. Sci. Technol. 2016, 6, 5304.
doi: 10.1039/C6CY00457A pmid: 28066540 |
| [86] |
Li, B.-W.; Wang, M.-Y.; Fang, S.; Liu, J.-Y. Organometallics 2019, 38, 2189.
doi: 10.1021/acs.organomet.9b00168 |
| [87] |
Testa, C.; Roger, J.; Scheib, S.; Fleurat-Lessard, P.; Hierso, J.-C. Adv. Synth. Catal. 2015, 357, 2913.
doi: 10.1002/adsc.201500321 |
| [88] |
Furuya, T.; Benitez, D.; Tkatchouk, E.; Strom, A. E.; Tang, P.; Goddard, W. A.; Ritter, T. J. Am. Chem. Soc. 2010, 132, 3793.
doi: 10.1021/ja909371t |
| [89] |
Cui, L.; Saeys, M. Chem. Cat. Chem. 2011, 3, 1060.
|
| [90] |
Katcher, M. H.; Norrby, P.-O.; Doyle, A. G. Organometallics 2014, 33, 2121.
doi: 10.1021/om401240p |
| [91] |
Mao, Y.-J.; Luo, G.; Hao, H.-Y.; Xu, Z.-Y.; Lou, S.-J.; Xu, D.-Q. Chem. Commun. 2019, 55, 14458.
doi: 10.1039/C9CC07726J |
| [92] |
Haines, B. E.; Xu, H.; Verma, P.; Wang, X.-C.; Yu, J.-Q.; Musaev, D. G. J. Am. Chem. Soc. 2015, 137, 9022.
doi: 10.1021/jacs.5b03410 |
| [93] |
Zhou, M.-J.; Yang, T.-L.; Dang, L. J. Org. Chem. 2016, 81, 1006.
doi: 10.1021/acs.joc.5b02571 |
| [94] |
Saito, H.; Yamamoto, K.; Sumiya, Y.; Liu, L.-J.; Nogi, K.; Maeda, S.; Yorimitsu, H. Chem. Asian J. 2020, 15, 2442.
doi: 10.1002/asia.202000591 |
| [95] |
Jaiswal, Y.; Kumar, Y.; Kumar, A. Org. Biomol. Chem. 2019, 17, 6809.
doi: 10.1039/c9ob01082c pmid: 31246220 |
| [96] |
Zhou, Y.-P.; Wang, M.-Y.; Fang, S.; Chen, Y.; Liu, J.-Y. RSC Adv. 2016, 6, 18300.
doi: 10.1039/C5RA27324B |
| [97] |
Tang, B.-C.; Lin, W.-X.; Chen, X.-L.; He, C.; Ma, J.-T.; Wu, Y.-D.; Lan, Y.; Wu, A.-X. Nat. Commun. 2020, 11, 5662.
doi: 10.1038/s41467-020-19508-z |
| [98] |
Garçon, M.; Mun, N. W.; White, A. J. P.; Crimmin, M. R. Angew. Chem., Int. Ed. 2021, 60, 6145.
doi: 10.1002/anie.202014960 |
| [99] |
Garçon, M.; White, A. J. P.; Crimmin, M. R. Chem. Commun. 2018, 54, 12326.
doi: 10.1039/C8CC06392C |
| [100] |
Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem.-Eur. J. 2010, 16, 2654.
doi: 10.1002/chem.200902374 |
| [101] |
Baudoin, O. Chem. Soc. Rev. 2011, 40, 4902.
doi: 10.1039/c1cs15058h |
| [102] |
Chen, F.-J.; Zhao, S.; Hu, F.; Chen, K.; Zhang, Q.; Zhang, S.-Q.; Shi, B.-F. Chem. Sci. 2013, 4, 4187.
doi: 10.1039/c3sc51993g |
| [103] |
Munz, D.; Meyer, D.; Strassner, T. Organometallics 2013, 32, 3469.
doi: 10.1021/om400232u |
| [104] |
Cundari, T. R.; Prince, B. M. J. Organomet. Chem. 2011, 696, 3982.
doi: 10.1016/j.jorganchem.2011.06.015 |
| [105] |
Prince, B. M. Comput. Theor. Chem. 2019, 1162, 112503.
doi: 10.1016/j.comptc.2019.112503 |
| [106] |
Wang, M.; Yang, Y.; Fan, Z.; Cheng, Z.; Zhu, W.; Zhang, A. Chem. Commun. 2015, 51, 3219.
doi: 10.1039/C4CC09576F |
| [107] |
Canty, A. J.; Ariafard, A.; Camasso, N. M.; Higgs, A. T.; Yates, B. F.; Sanford, M. S. Dalton. Trans. 2017, 46, 3742.
doi: 10.1039/C7DT00096K |
| [108] |
Buettner, C. S.; Willcox, D.; Chappell, B. G. N.; Gaunt, M. J. Chem. Sci. 2019, 10, 83.
doi: 10.1039/C8SC03434F |
| [109] |
Pan, J.; Su, M.; Buchwald, S. L. Angew. Chem., Int. Ed. 2011, 50, 8647.
doi: 10.1002/anie.201102880 |
| [110] |
Iglesias, Á.; Álvarez, R.; de Lera, Á. R.; Muñiz, K. Angew. Chem., Int. Ed. 2012, 51, 2225.
doi: 10.1002/anie.201108351 |
| [111] |
McNally, A.; Haffemayer, B.; Collins, B. S. L.; Gaunt, M. J. Nature 2014, 510, 129.
doi: 10.1038/nature13389 |
| [112] |
Zhang, Q.; Yu, H.; Fu, Y. Sci. China Chem. 2015, 58, 1316.
doi: 10.1007/s11426-015-5360-7 |
| [113] |
Zhang, Y.; Qi, Z.-H.; Ruan, G.-Y.; Zhang, Y.; Liu, W.; Wang, Y. RSC Adv. 2015, 5, 71586.
doi: 10.1039/C5RA11488H |
| [114] |
Smalley, A. P.; Gaunt, M. J. J. Am. Chem. Soc. 2015, 137, 10632.
doi: 10.1021/jacs.5b05529 pmid: 26247373 |
| [115] |
Zakrzewski, J.; Smalley, A. P.; Kabeshov, M. A.; Gaunt, M. J. Lapkin, A. A. Angew. Chem., Int. Ed. 2016, 55, 8878.
doi: 10.1002/anie.201602483 |
| [116] |
Duarte, F. J. S.; Poli, G.; Calhorda, M. J. ACS Catal. 2016, 6, 1772.
doi: 10.1021/acscatal.5b02091 |
| [117] |
Tong, H.-R.; Zheng, W.; Lv, X.; He, G.; Liu, P.; Chen, G. ACS Catal. 2020, 10, 114.
doi: 10.1021/acscatal.9b04768 |
| [118] |
Sun, H.; Zhang, Y.; Chen, P.; Wu, Y.-D.; Zhang, X.; Huang, Y. Adv. Synth. Catal. 2016, 358, 1946.
doi: 10.1002/adsc.201600015 |
| [119] |
Chen, Y.-Q.; Singh, S.; Wu, Y.; Wang, Z.; Hao, W.; Verma, P.; Qiao, J. X.; Sunoj, R. B.; Yu, J.-Q. J. Am. Chem. Soc. 2020, 142, 9966.
doi: 10.1021/jacs.9b13537 |
| [120] |
Giri, R.; Lan, Y.; Liu, P.; Houk, K. N.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 14118.
doi: 10.1021/ja304643e |
| [121] |
Deb, A.; Singh, S.; Seth, K.; Pimparkar, S.; Bhaskararao, B.; Guin, S.; Sunoj, R. B.; Maiti, D. ACS Catal. 2017, 7, 8171.
doi: 10.1021/acscatal.7b03056 |
| [122] |
He, J.; Shao, Q.; Wu, Q.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 3344.
doi: 10.1021/jacs.6b13389 |
| [123] |
Xing, Y.-Y.; Liu, J.-B.; Sun, Q.-M.; Sun, C.-Z.; Huang, F.; Chen, D.-Z. J. Org. Chem. 2019, 84, 10690.
doi: 10.1021/acs.joc.9b01227 |
| [1] | 付雅彤, 孙超凡, 张丹, 金成国, 陆居有. 巢式-碳硼烷硼氢键官能化反应研究进展[J]. 有机化学, 2024, 44(2): 438-447. |
| [2] | 张剑, 梁万洁, 杨艺, 闫法超, 刘会. 联烯胺化合物的区域选择性双官能团化[J]. 有机化学, 2024, 44(2): 335-348. |
| [3] | 陈宛婷, 钟雄威, 邢佳乐, 吴昌书, 高杨. C—N轴手性化合物的不对称催化合成研究进展[J]. 有机化学, 2024, 44(2): 349-377. |
| [4] | 孟宪强, 杨艺, 梁万洁, 王靖涛, 张荣葵, 刘会. 钯催化联烯胺区域选择性芳基酚氧化反应[J]. 有机化学, 2024, 44(1): 224-231. |
| [5] | 王婧怡, 刘金羽, 陈东升, 陈华燕, 谢欣, 南发俊. 新型选择性半胱氨酰白三烯受体1 (CysLT1R)拮抗剂的设计合成及构效关系研究[J]. 有机化学, 2024, 44(1): 259-276. |
| [6] | 张莹珍, 江丹丹, 李娟华, 王菁菁, 刘昆明, 刘晋彪. 高选择性硒代半胱氨酸荧光探针的构建策略及成像[J]. 有机化学, 2024, 44(1): 41-53. |
| [7] | 张俊杰, 徐学涛. (S)-(–)-Xylopinine和(S)-(+)-Laudanosine的不对称合成[J]. 有机化学, 2023, 43(9): 3297-3303. |
| [8] | 王兢睿, 冯永奎, 王能中, 黄年玉, 姚辉. 钯催化立体选择性合成硝基烷类β-碳糖苷[J]. 有机化学, 2023, 43(9): 3216-3225. |
| [9] | 王玉超, 刘晋彪, 何智涛. 钯催化共轭二烯的不对称氢官能团化[J]. 有机化学, 2023, 43(8): 2614-2627. |
| [10] | 范威. O2促进下五元环烯胺的C—H亚胺化[J]. 有机化学, 2023, 43(7): 2492-2498. |
| [11] | 户晓兢, 郭斐翔, 朱润青, 周柄棋, 张涛, 房立真. 对烷氧基酚的合成及其去芳构化后的合成应用[J]. 有机化学, 2023, 43(6): 2239-2244. |
| [12] | 蒋旺, 史壮志. 芳烃间/对位选择性碳氢硼化反应研究进展[J]. 有机化学, 2023, 43(5): 1691-1705. |
| [13] | 纪健, 刘进华, 管丛, 陈绪文, 赵芸, 刘顺英. 原位生成的磺酸催化N-磺酰基-1,2,3-三氮唑与醇偶联高区域选择性合成N2-取代1,2,3-三氮唑[J]. 有机化学, 2023, 43(3): 1168-1176. |
| [14] | 向勋, 何照林, 董秀琴. 钯和手性磷酸协同催化高效构建手性分子的研究进展[J]. 有机化学, 2023, 43(3): 791-808. |
| [15] | 郭萍, 周勇, 赵杰. 多取代烯烃的Z∶E高选择性合成制备[J]. 有机化学, 2023, 43(3): 855-872. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
