有机化学 ›› 2026, Vol. 46 ›› Issue (2): 507-514.DOI: 10.6023/cjoc202507040 上一篇 下一篇
研究论文
陈梓桐a, 王力为a,*( ), 沈晓b, 戚孝天a,*()
), 沈晓b, 戚孝天a,*()
收稿日期:2025-07-29
修回日期:2025-10-03
发布日期:2025-11-05
通讯作者:
王力为, 戚孝天
基金资助:
Zitong Chena, Liwei Wanga,*(), Xiao Shenb, Xiaotian Qia,*()
Received:2025-07-29
Revised:2025-10-03
Published:2025-11-05
Contact:
Liwei Wang, Xiaotian Qi
Supported by:文章分享
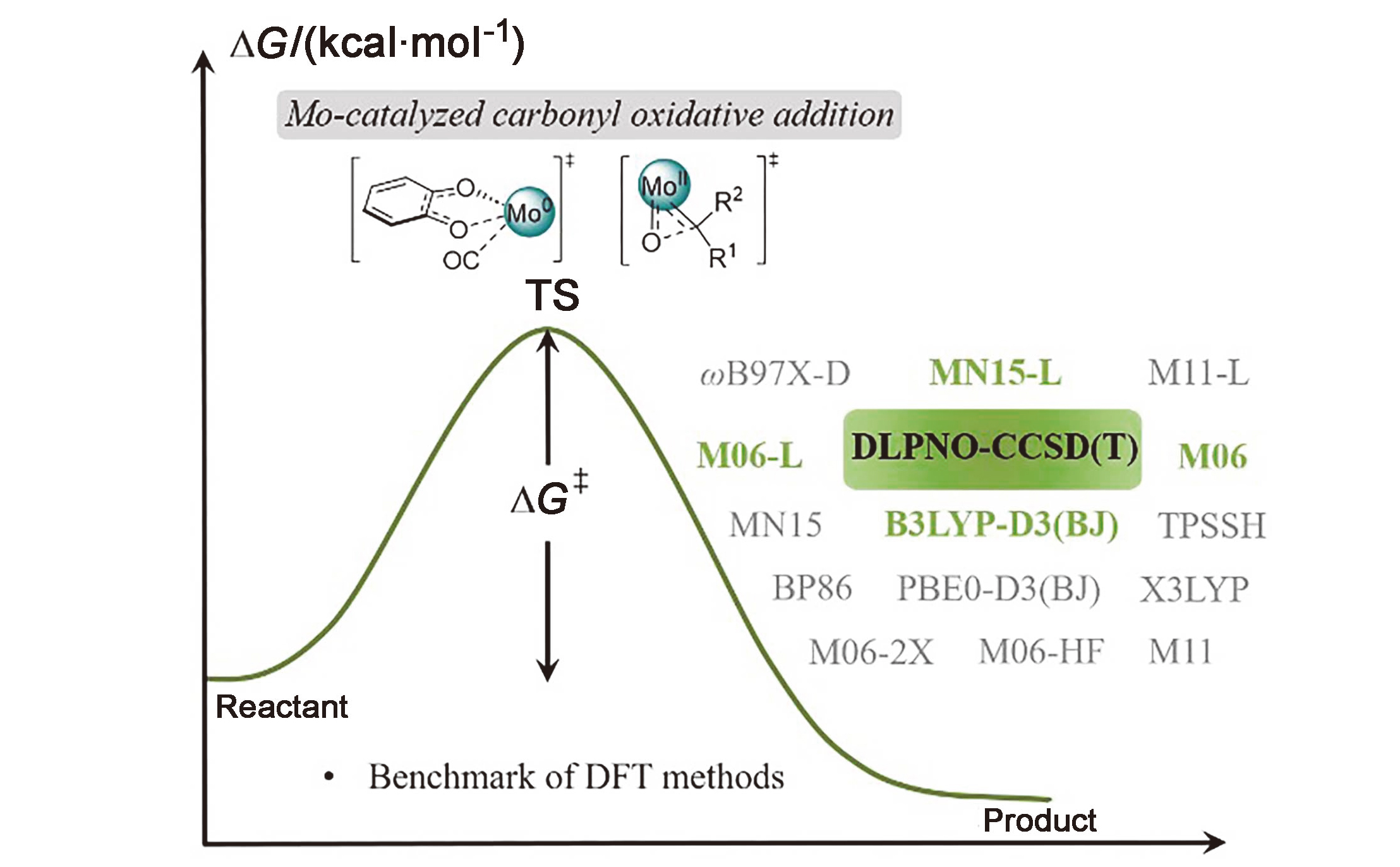
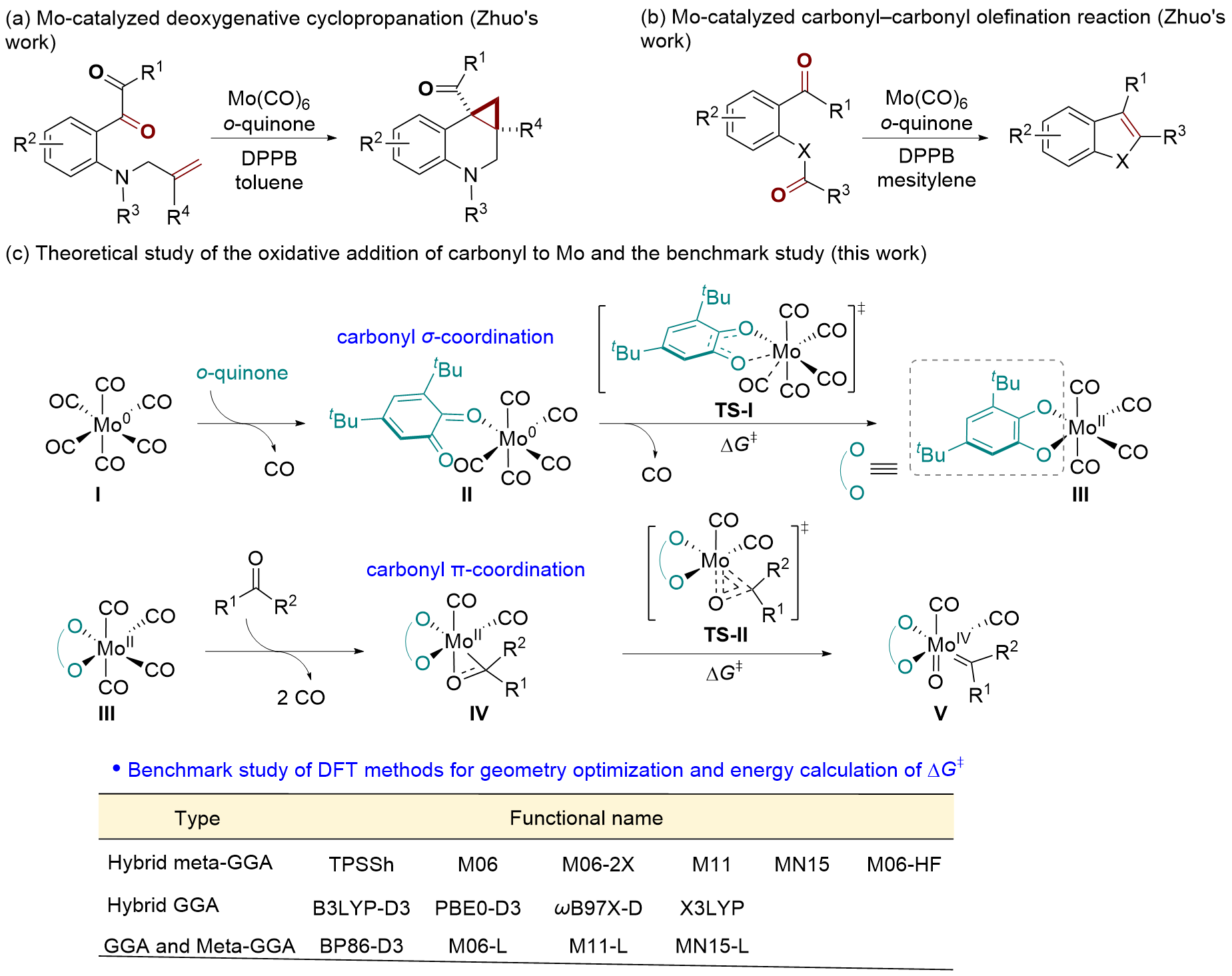
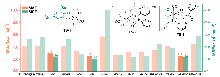
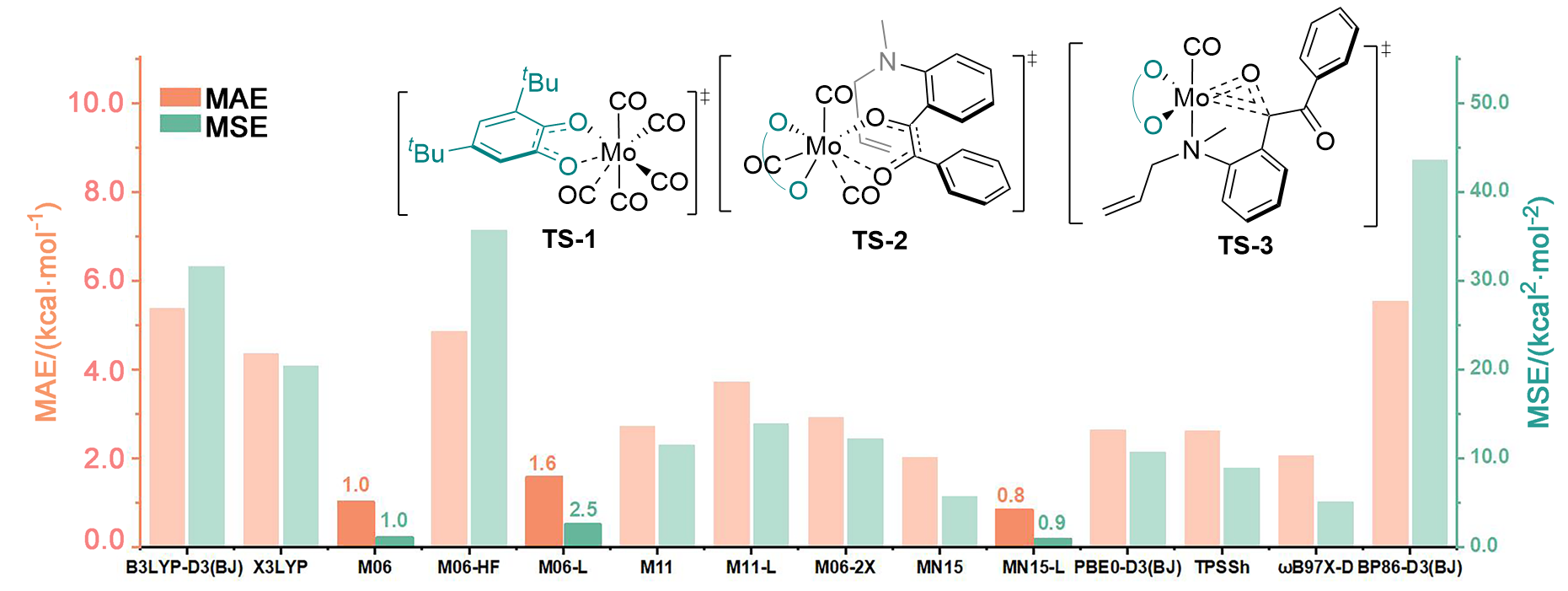
在钼化学中, 邻苯醌或1,2-二羰基化合物对钼的氧化加成被广泛应用于钼催化的碳碳成键反应. 其中羰基对零价或者二价钼的协同氧化加成是钼催化的关键基元步骤. 关于该基元步骤的密度泛函理论(DFT)计算研究相对较少, 特别是对泛函的选取缺少参考. 因此, 选择了14种密度泛函计算钼催化的羰基氧化加成过程, 评估了不同泛函对于结构优化和能量计算的准确性. 平均绝对误差(MAE)和平均平方误差(MSE)分析表明, B3LYP-D3(BJ)、TPSSh和ωB97X-D泛函在结构优化方面具有较好的表现. 此外, 基于B3LYP-D3(BJ)泛函在气相中优化得到的结构, 使用M06、M06-L和MN15-L泛函进行能量计算的结果与DLPNO-CCSD(T)泛函计算得到的数据更为接近, 特别是MN15-L的表现最优, 具有最小的MAE和MSE.
陈梓桐, 王力为, 沈晓, 戚孝天. 钼催化羰基氧化加成步骤的密度泛函理论(DFT)方法评估研究[J]. 有机化学, 2026, 46(2): 507-514.
Zitong Chen, Liwei Wang, Xiao Shen, Xiaotian Qi. A Benchmark Study of Density Functional Theory (DFT) Methods for Mo-Catalyzed Carbonyl Oxidative Addition[J]. Chinese Journal of Organic Chemistry, 2026, 46(2): 507-514.
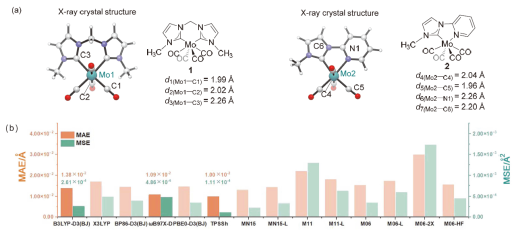

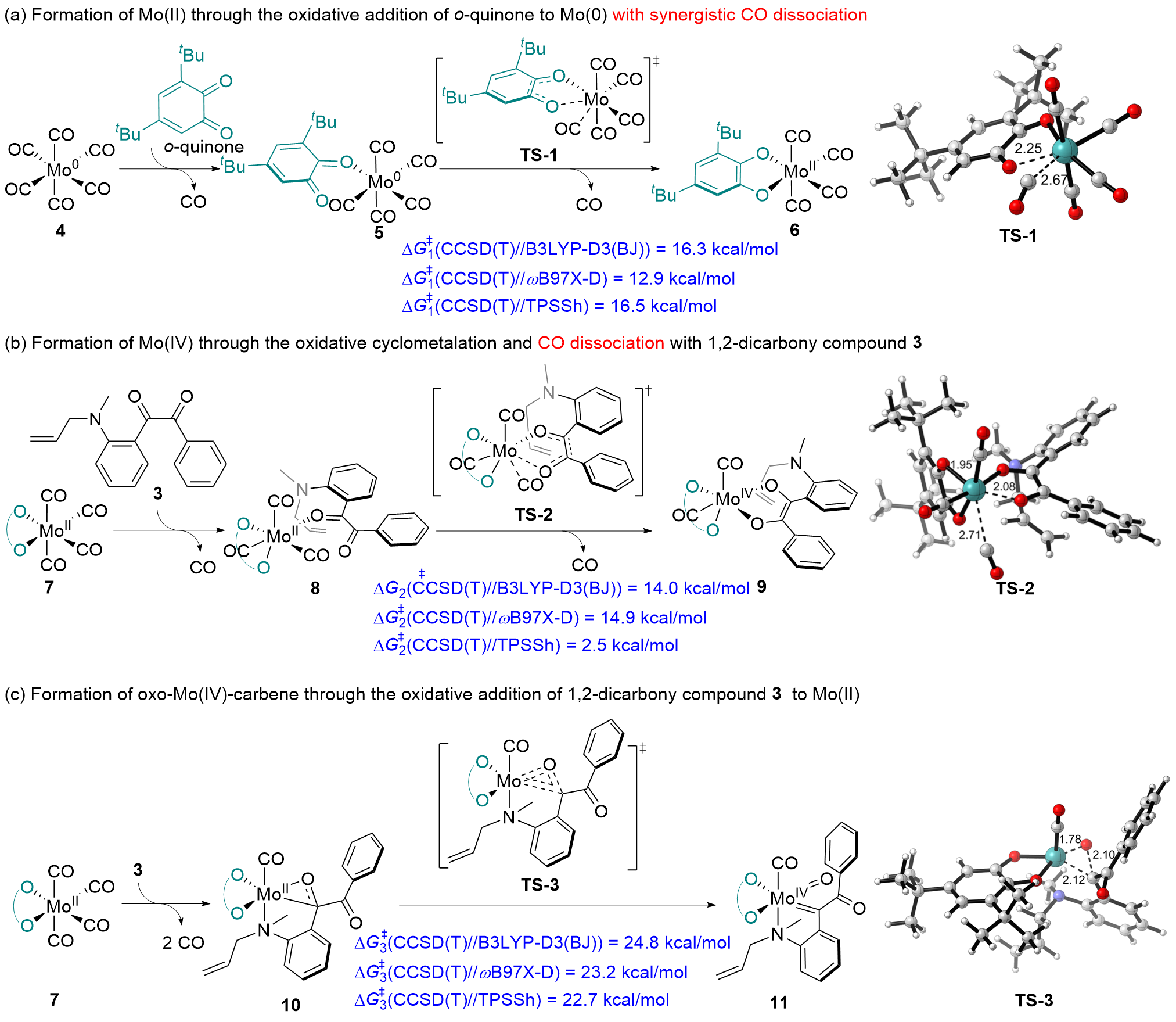
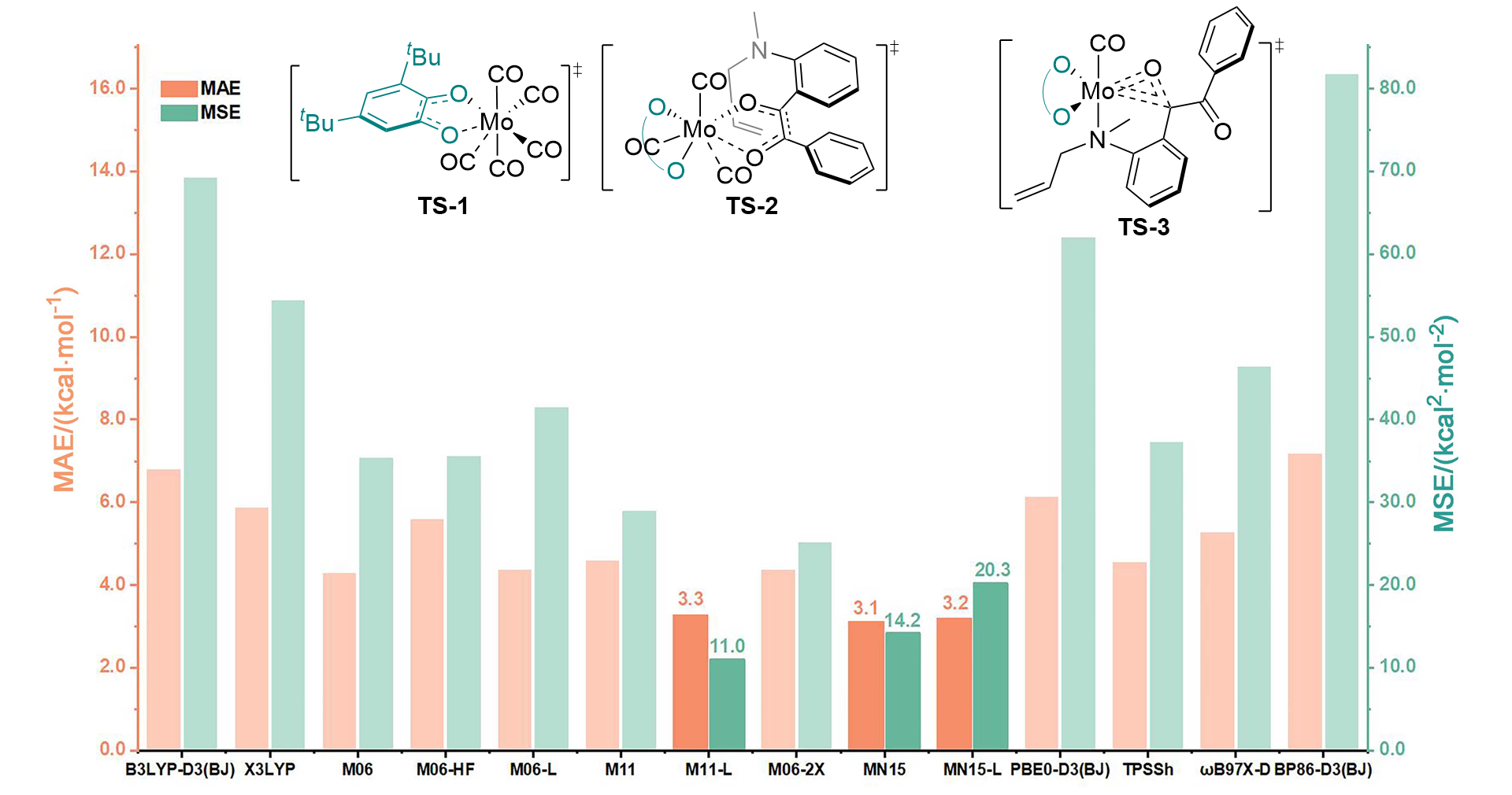
| [15] |
(a)
doi: 10.1038/s41467-019-12467-0 pmid: 31582755 |
|
(b)
doi: 10.1146/physchem.2017.68.issue-1 pmid: 31582755 |
|
| [16] |
doi: 10.1063/5.0128508 |
| [17] |
doi: 10.1103/PhysRevB.102.035129 |
| [18] |
|
| [19] |
doi: 10.1021/acs.jctc.8b00288 |
| [20] |
Frisch, M. J. T. G. W.;
|
| [21] |
(a)
doi: 10.1002/wcms.v2.1 |
|
(b)
|
|
|
(c)
doi: 10.1063/5.0004608 |
|
| [1] |
(a)
doi: 10.1093/advances/nmx001 pmid: 29767695 |
|
(b)
doi: 10.6023/cjoc201707017 pmid: 29767695 |
|
|
(胡传峰, 周建豪, 黄志达, 傅惠惠, 彭新华, 有机化学, 2018, 38, 486.)
doi: 10.6023/cjoc201707017 pmid: 29767695 |
|
|
(c)
pmid: 29767695 |
|
|
(宋礼成, 罗春成, 有机化学, 2001, 21, 1009.)
pmid: 29767695 |
|
| [2] |
doi: 10.1016/j.ccr.2011.01.027 |
| [3] |
(a)
pmid: 12114025 |
|
(b)
doi: 10.1016/s0968-0004(02)02107-2 pmid: 12114025 |
|
|
(c)
pmid: 12114025 |
|
| [4] |
(a)
doi: 10.1038/nchem.2011 pmid: 25054948 |
|
(b)
doi: 10.1002/chem.v25.1 pmid: 25054948 |
|
| [5] |
(a)
doi: 10.1002/anie.v45:23 pmid: 27126041 |
|
(b)
doi: 10.1126/science.aaf4622 pmid: 27126041 |
|
| [22] |
doi: 10.1016/j.trechm.2020.02.005 |
| [23] |
doi: 10.1103/PhysRevB.33.8822 |
| [24] |
(a)
doi: 10.1021/j100096a001 |
|
(b)
doi: 10.1063/1.464913 |
|
| [25] |
doi: 10.1063/1.478522 |
| [26] |
doi: 10.1016/j.cplett.2015.12.069 |
| [27] |
doi: 10.1063/1.3382344 |
| [28] |
doi: 10.1039/b810189b |
| [29] |
doi: 10.1063/1.2370993 |
| [30] |
doi: 10.1021/jz201525m |
| [31] |
doi: 10.1021/acs.jctc.5b01082 |
| [5] |
(c)
doi: 10.1021/jacs.3c10430 pmid: 27126041 |
| [6] |
doi: 10.1021/jacs.7b03070 pmid: 28414434 |
| [7] |
doi: 10.1002/chem.v10:2 |
| [8] |
(a)
doi: 10.1021/cr400443z pmid: 34045715 |
|
(b)
doi: 10.1038/s41557-021-00701-6 pmid: 34045715 |
|
|
(c)
doi: 10.1002/anie.v53.43 pmid: 34045715 |
|
|
(d)
doi: 10.1039/D0CS00923G pmid: 34045715 |
|
| [9] |
(a)
doi: 10.1007/s12274-022-5277-3 |
|
(b)
doi: 10.1016/j.nanoen.2019.04.060 |
|
| [32] |
doi: 10.1007/s00214-007-0310-x |
| [33] |
doi: 10.1021/jz201170d |
| [34] |
doi: 10.1039/C6SC00705H |
| [35] |
(a)
doi: 10.1103/PhysRevLett.91.146401 |
|
(b)
doi: 10.1063/1.1795692 |
|
| [36] |
doi: 10.1073/pnas.0308730100 |
| [37] |
doi: 10.1063/5.0206533 |
| [38] |
(a)
doi: 10.1039/b508541a |
|
(b)
doi: 10.1039/b515623h |
|
| [39] |
doi: 10.1021/jp810292n |
| [10] |
doi: 10.1002/anie.v60.28 |
| [11] |
doi: 10.1039/D3QO00567D |
| [12] |
doi: 10.6023/cjoc202405018 |
|
(孙庆浩, 鲍晓光, 有机化学, 2024, 44, 3518.)
doi: 10.6023/cjoc202405018 |
|
| [13] |
(a)
doi: 10.1002/advs.v11.47 |
|
(b)
doi: 10.1021/acscatal.6b02523 |
|
| [14] |
(a)
doi: 10.1007/s11051-017-4072-7 pmid: 29206033 |
|
(b)
doi: 10.1021/acs.inorgchem.7b02133 pmid: 29206033 |
|
| [40] |
(a)
doi: 10.1002/jcc.v29:2 |
|
(b)
doi: 10.1007/s00214-007-0250-5 |
|
| [41] |
|
| [42] |
doi: 10.1021/acscatal.1c02956 |
| [1] | 令小鹏, 陶绍平, 林奇, 史兵兵, 姚虹, 魏太保, 陈进发. 基于π-共轭柱[5]芳烃的荧光传感器用于L-精氨酸识别[J]. 有机化学, 2025, 45(7): 2480-2485. |
| [2] | 王涛, 陶晟, 陈飞, 杜智宏, 薄春博, 李敏, 刘宁. 双功能钼配合物催化二氧化碳和环氧化物制备环状碳酸酯[J]. 有机化学, 2025, 45(12): 4354-4361. |
| [3] | 孙庆浩, 鲍晓光. 钼催化芳香醛脱氧偶联反应机制的理论研究[J]. 有机化学, 2024, 44(11): 3518-3525. |
| [4] | 陈学伟, 于方彩, 田传洪. 1,1'-亚甲基二咪唑鎓多氢键供体催化剂促进常压下CO2与环氧化物的环加成反应[J]. 有机化学, 2024, 44(10): 3198-3205. |
| [5] | 李建文, 王涛, 陶晟, 陈飞, 李敏, 刘宁. SBA-15负载的N-杂环卡宾-吡啶钼配合物在二氧化碳转化制备环状碳酸酯中的应用[J]. 有机化学, 2024, 44(10): 3213-3222. |
| [6] | 刘婷婷, 胡宇才, 沈安. 亚胺配体协同氮杂环卡宾钯配合物催化碳碳偶联反应的作用机制[J]. 有机化学, 2023, 43(2): 622-628. |
| [7] | 李英俊, 赵月, 靳焜, 高立信, 盛丽, 刘雪洁, 杨鸿境, 林乐弟, 李佳. 含咔唑/苯并咪唑环的2,5-二取代-1,3,4-噻二唑酰胺衍生物的合成及对PTP1B/TCPTP抑制活性的评价[J]. 有机化学, 2019, 39(9): 2599-2608. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
