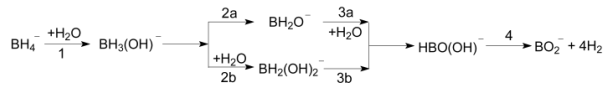
| [1] Martelli, P.; Caputo, R.; Remhof, A.; Mauron, P.; Borgschulte, A.; Zu?ttel, A. J. Phys. Chem. C 2010, 114, 7173. [2] Shang, Y.; Chen, R. Energy Fuels 2006, 20, 2142. [3] Kumar, R. S.; Kim, E.; Cornelius, A. L. J. Phys. Chem. C 2008, 112, 8452. [4] Churikov, A. V.; Zapsis, K. V.; Khramkov, V. V.; Churikov, M. A.; Smotrov, M. P.; Kazarinov, I. A. J. Chem. Eng. Data 2010, 56, 9. [5] Fang, F.; Li, Y.; Song, Y.; Sun, D.; Zhang, Q.; Ouyang, L.; Zhu, M. J. Phys. Chem. C 2011, 115, 13528. [6] Laversenne, L.; Goutaudier, C.; Chiriac, R.; Sigala, C.; Bonnetot, B. J. Therm. Anal. Calorim. 2008, 94, 785. [7] Groshens, T. J.; Hollins, R. A. Chem. Commun. 2009, 3089. [8] Li, L.; Li, S.; Tan, Y.; Tang, Z.; Cai, W.; Guo, Y.; Li, Q.; Yu, X. J. Phys. Chem. C 2012, 116, 14218. [9] Schulz, R.; Huot, J.; Liang, G.; Boily, S.; Lalande, G.; Denis, M. C.; Dodelet, J. P. Mater. Sci. Eng. A 1999, 267, 240. [10] Gardiner, J. A.; Collat, J. W. J. Am. Chem. Soc. 1965, 87, 1692. [11] Sousa, T.; Fernandes, V. R.; Pinto, P. J. R.; Slavkov, Y.; Bosukov, L.; Rangel, C. M. Chem. Eng. Sci. 2012, 84, 70. [12] Xu, D.; Zhao, L.; Dai, P.; Ji, S. J. Nat. Gas Chem. 2012, 21, 488. [13] Marreroalfonso, E.; Gray, J.; Davis, T.; Matthews, M. Int. J. Hydrogen Energy 2007, 32, 4723. [14] Guella, G.; Zanchetta, C.; Patton, B.; Miotello, A. J. Phys. Chem. B 2006, 110, 17024. [15] Politzer, P.; Lane, P. J. J. Mol. Struct. 1998, 454, 229. [16] Hehre, W. J.; Radom, L.; Schleyer, P. V. R.; Pople, J. A. Ab initio Molecular Orbital Theory, Wiley, New York, 1986, p. 38. [17] Frisch, M. J.; Pople, J. A.; Binkley, J. S. J. Chem. Phys. 1984, 80, 3265. [18] Hehre, W. J.; Radom, L.; Schleyer, P. V. R.; Pople, J. A. Ab initio Molecular Orbital Theory, Wiley, New York, 1986, p. 86. [19] Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, revision B.01, Gaussian, Inc., Wallingford, CT, 2010. [20] Mo, L.-X.; Zeng, Y.-L.; Zhang, X.-Y.; Zheng, S.-J.; Meng, L.-P. Acta Phys. Chim. Sin. 2007, 23, 120. (默丽欣, 曾艳丽, 张雪英, 郑世钧, 孟令鹏, 物理化学学报, 2007, 23, 120.) [21] Diakov, V.; Diwan, M.; Shafirovich, E.; Varma, A. Chem. Eng. Sci. 2007, 62, 5586. [22] Liu, B. H.; Li, Z. P. J. Power Sources 2009, 187, 527. [23] Crisafulli, C.; Scirè, S.; Salanitri, M.; Zito, R.; Calamia, S. Int. J. Hydrogen Energy 2011, 36, 3817. |