化学学报 ›› 2022, Vol. 80 ›› Issue (6): 781-787.DOI: 10.6023/A22010056 上一篇 下一篇
研究论文
王珞聪, 李哲伟, 岳彩巍, 张培焕, 雷鸣*( ), 蒲敏*()
), 蒲敏*()
投稿日期:2022-01-30
发布日期:2022-07-07
通讯作者:
雷鸣, 蒲敏
基金资助:
Luocong Wang, Zhewei Li, Caiwei Yue, Peihuan Zhang, Ming Lei(), Min Pu()
Received:2022-01-30
Published:2022-07-07
Contact:
Ming Lei, Min Pu
Supported by:文章分享

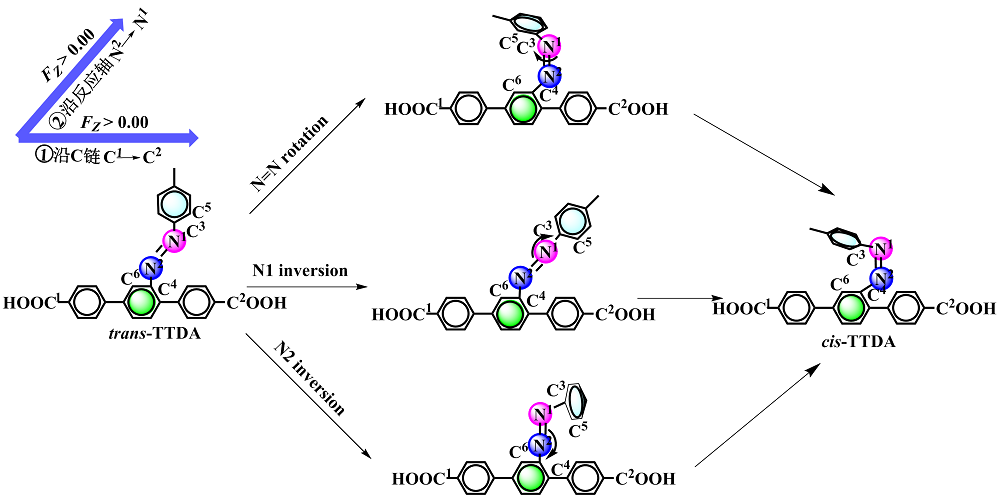
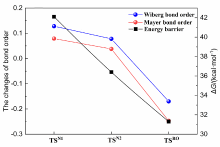
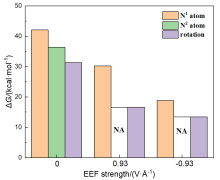
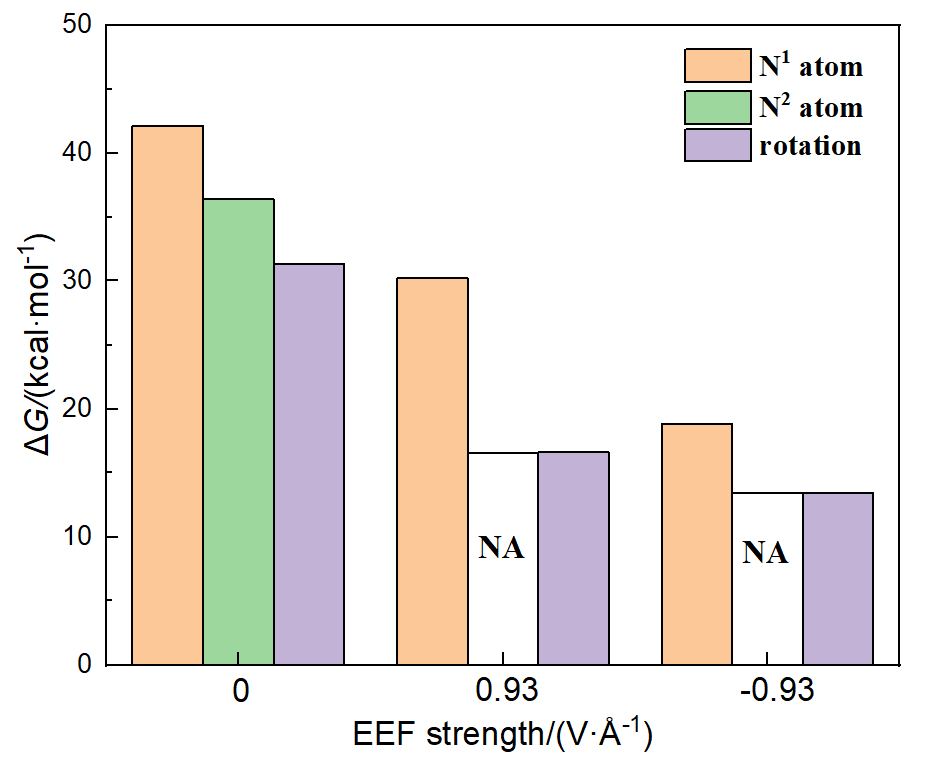
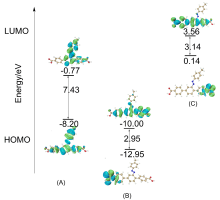
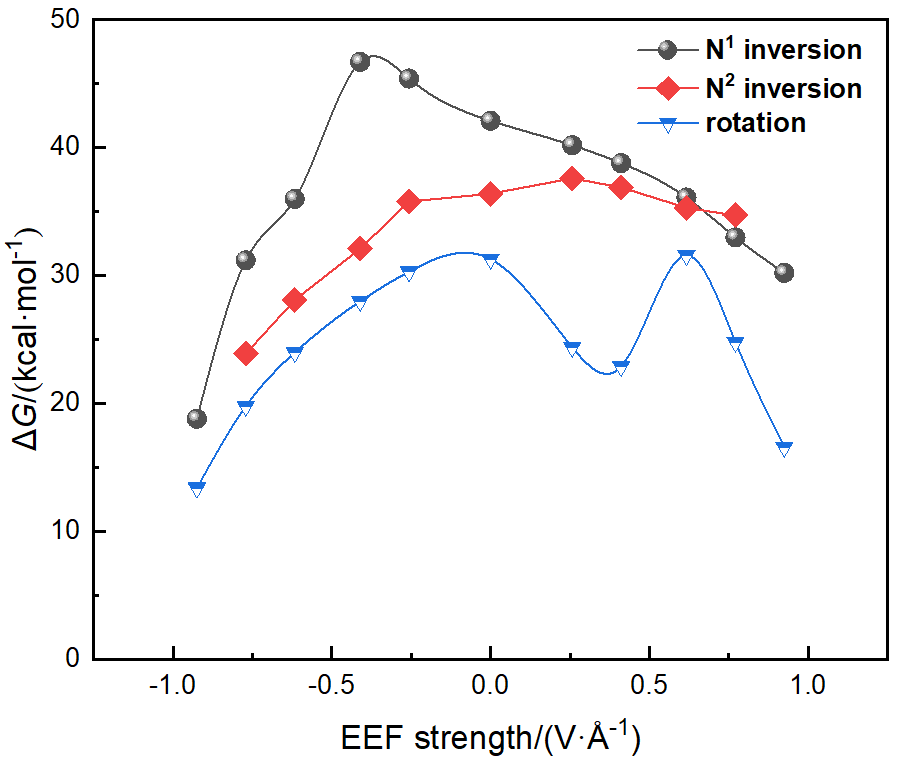
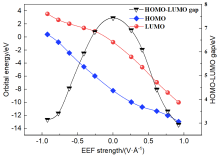
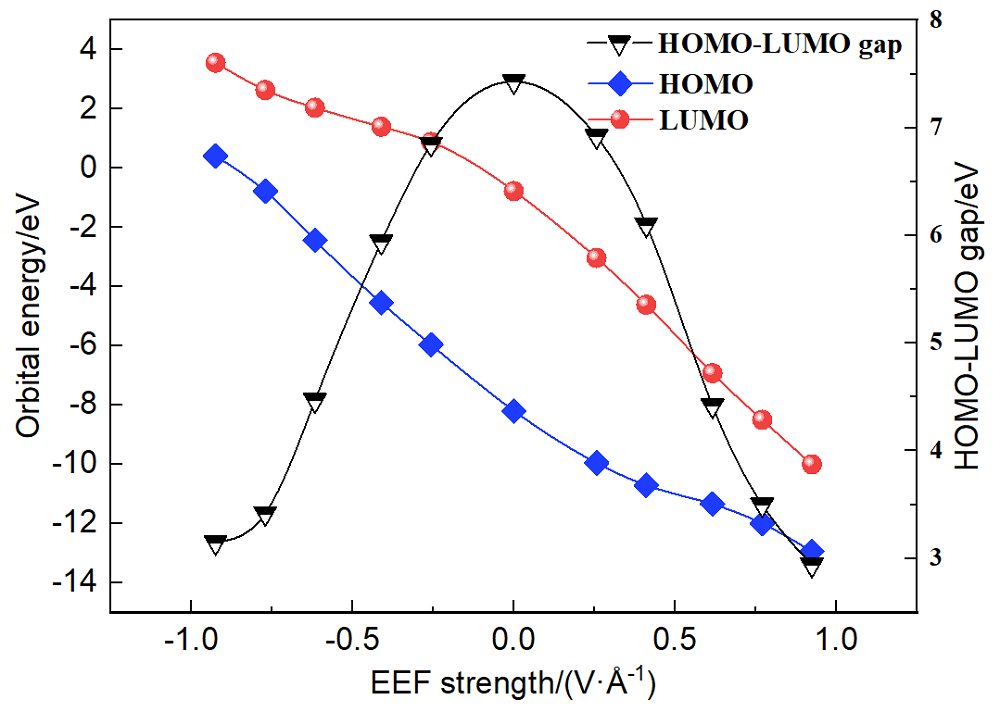
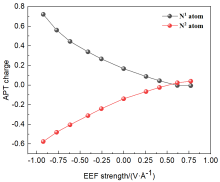
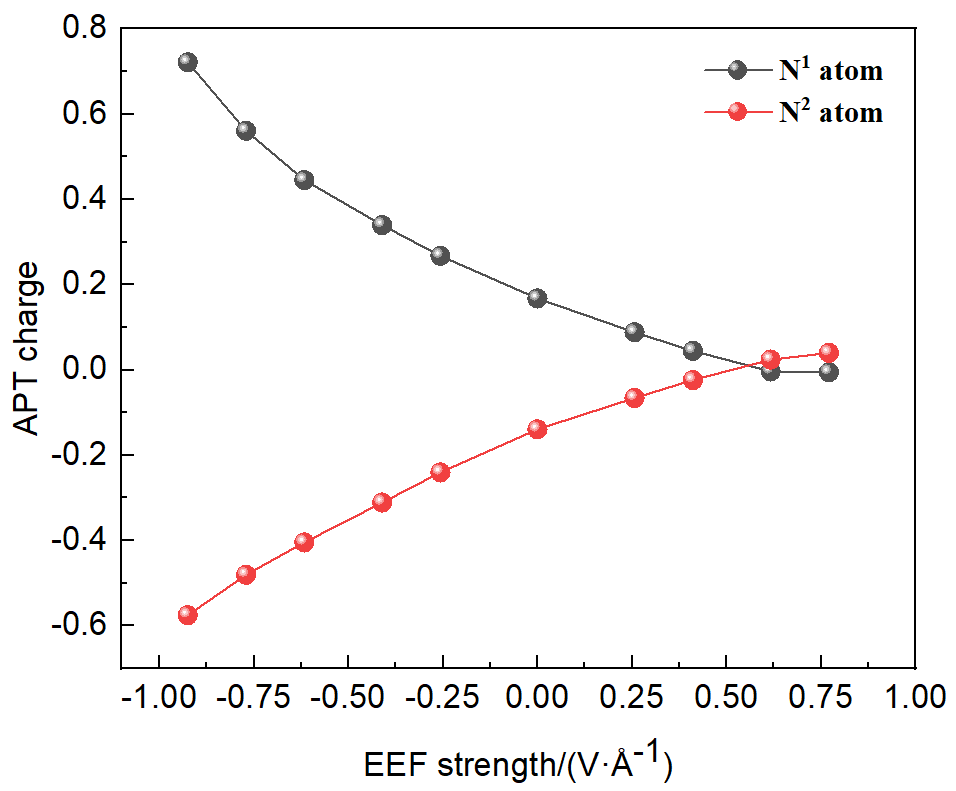
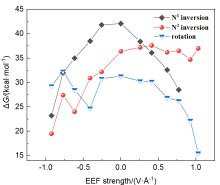
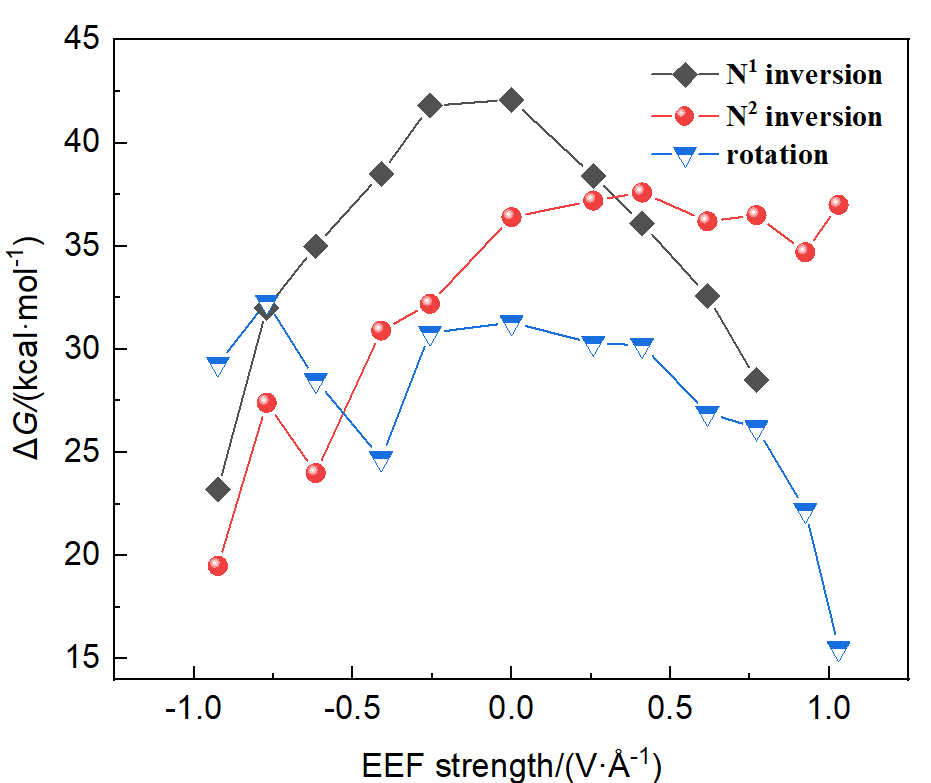
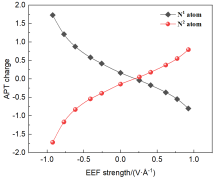
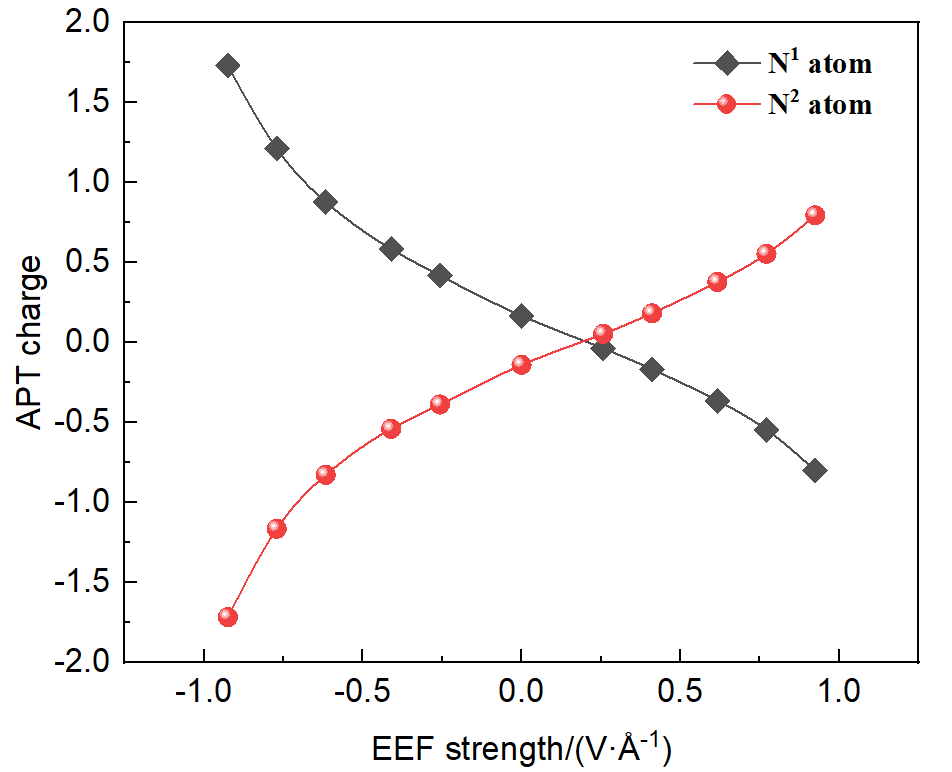
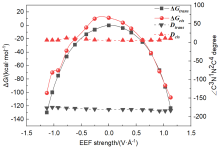
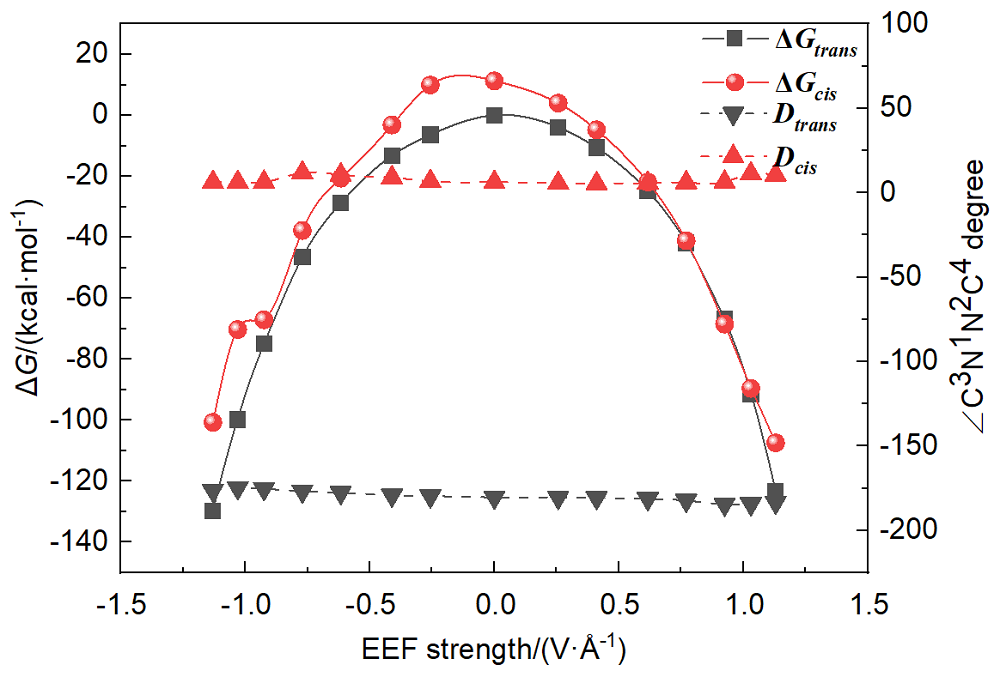
本工作采用密度泛函理论(DFT)方法计算研究了不同电场强度下偶氮苯衍生物2'-对甲苯偶氮基-1,1':4,4'-三苯基- 4,4''二羧酸(TTDA)顺反异构化反应的机理. TTDA经过C—N1=N2角度顺反异构化过程存在三种可能的异构化模式 (N=N偶氮基团中与大取代基相连的N原子称为N2, 与小取代基相连的N原子称为N1), 绕N1或N2原子的反转和绕N1=N2键旋转. 计算结果表明, 加入沿z轴的电场(以三联苯侧链C1→C2方向为z轴正方向), 旋转路径为反应最优路径. 此外, 还研究了沿N=N键方向加入电场(以N2→N1方向为z轴正方向), 在电场强度Fz=0.00 V•Å-1时, N1反转路径能垒较N2反转路径高. 当–0.62 V•Å-1<Fz≤0.93 V•Å-1时, 旋转路径为优势路径. 当加入沿z轴的反向电场–0.93 V•Å-1≤Fz≤–0.62 V•Å-1时, N2反转为优势路径.
王珞聪, 李哲伟, 岳彩巍, 张培焕, 雷鸣, 蒲敏. 电场下偶氮苯衍生物分子顺反异构化反应机理的理论研究[J]. 化学学报, 2022, 80(6): 781-787.
Luocong Wang, Zhewei Li, Caiwei Yue, Peihuan Zhang, Ming Lei, Min Pu. Theoretical Study on the Isomerization Mechanism of Azobenzene Derivatives under Electric Field[J]. Acta Chimica Sinica, 2022, 80(6): 781-787.







| [1] |
Helal, W.; Bories, B.; Evangelisti, S.; Leininger, T.; Maynau, D. Computational Science and Its Applications, Springer, Berlin, Heidelberg, 2006, pp. 744-751.
|
| [2] |
Xiang, D.; Wang, X.; Jia, C.; Lee, T.; Guo, X. Chem. Rev. 2016, 116, 4318.
doi: 10.1021/acs.chemrev.5b00680 pmid: 26979510 |
| [3] |
Jia, C.; Migliore, A.; Xin, N.; Huang, S.; Wang, J.; Yang, Q.; Wang, S.; Chen, H.; Wang, D.; Feng, B. Science 2016, 352, 1443.
doi: 10.1126/science.aaf6298 |
| [4] |
zhang, J. L.; zhong, J. Q.; Lin, J. D.; Hu, W. P.; Wu, K.; Xu, G. Q.; Wee, A. T.; Chen, W. Chem. Soc. Rev. 2015, 44, 2998.
doi: 10.1039/C4CS00377B |
| [5] |
Jia, C.; Migliore, A.; Xin, N.; Huang, S.; Guo, X. Science 2016, 352, 1443.
doi: 10.1126/science.aaf6298 |
| [6] |
Atesci, H.; Kaliginedi, V.; Celis Gil, J. A.; Ozawa, H.; Thijssen, J. M.; Broekmann, P.; Haga, M.-A.; van der Molen, S. J. Nat. Nanotechnol. 2018, 13, 117.
doi: 10.1038/s41565-017-0016-8 |
| [7] |
Perrin, M. L.; Burzurí, E.; zant, H. Chem. Soc. Rev. 2015, 44, 902.
doi: 10.1039/C4CS00231H |
| [8] |
Song, H.; Kim, Y.; Jang, Y. H.; Jeong, H.; Reed, M. A.; Lee, T. Nature 2009, 462, 1039.
doi: 10.1038/nature08639 |
| [9] |
Quek, S. Y.; Kamenetska, M.; Steigerwald, M. L.; Choi, H. J.; Louie, S. G.; Hybertsen, M. S.; Neaton, J. B.; Venkataraman, L. Nat. Nanotechnol. 2009, 4, 230.
doi: 10.1038/nnano.2009.10 |
| [10] |
Perrin, M. L.; Verzijl, C. J. O.; Martin, C. A.; Shaikh, A. J.; Eelkema, R.; van Esch, J. H.; van Ruitenbeek, J. M.; Thijssen, J. M.; van der zant, H. S. J.; Dulić, D. Nat. Nanotechnol. 2013, 8, 282.
doi: 10.1038/nnano.2013.26 |
| [11] |
Su, T. A.; Li, H.; Steigerwald, M. L.; Venkataraman, L.; Nuckolls, C. Nat. Chem. 2015, 7, 215.
doi: 10.1038/nchem.2180 |
| [12] |
Kim, Y.; Jeong, W.; Kim, K.; Lee, W.; Reddy, P. Nat. Nanotechnol. 2014, 9, 881.
doi: 10.1038/nnano.2014.209 |
| [13] |
Reddy, P.; Jang, S.-Y.; Segalman Rachel, A.; Majumdar, A. Science 2007, 315, 1568.
doi: 10.1126/science.1137149 |
| [14] |
Reecht, G.; Scheurer, F.; Speisser, V.; Dappe, Y. J.; Mathevet, F.; Schull, G. Phys. Rev. Lett. 2014, 112, 047403.
doi: 10.1103/PhysRevLett.112.047403 |
| [15] |
Reecht, G.; Scheurer, F.; Speisser, V.; Dappe, Y. J.; Mathevet, F.; Schull, G. Phys. Rev. Lett. 2014, 112, 047403.
doi: 10.1103/PhysRevLett.112.047403 |
| [16] |
Thiele, S.; Balestro, F.; Ballou, R.; Klyatskaya, S.; Ruben, M.; Wernsdorfer, W. Science 2014, 344, 1135.
doi: 10.1126/science.1249802 |
| [17] |
Natterer, F. D.; Yang, K.; Paul, W.; Willke, P.; Choi, T.; Greber, T.; Heinrich, A. J.; Lutz, C. P. Nature 2017, 543, 226.
doi: 10.1038/nature21371 |
| [18] |
Liu, Z.; Ren, S.; Guo, X. Molecular-Scale Electronics 2019, 173.
|
| [19] |
Yin, X.; Zang, Y.; Zhu, L.; Low, J. Z.; Liu, Z. F.; Cui, J.; Neaton, J. B.; Venkataraman, L.; Campos, L. M. Sci. Adv. 2017, 3, eaao2615.
doi: 10.1126/sciadv.aao2615 |
| [20] |
Liu, X.; Qin, L.; Zhan, Y.; Chen, M.; Yu, Y. Acta Chim. Sinica 2020, 78, 478. (in Chinese)
doi: 10.6023/A20040103 |
|
(刘晓珺, 秦朗, 詹媛媛, 陈萌, 俞燕蕾, 化学学报, 2020, 78, 478.)
doi: 10.6023/A20040103 |
|
| [21] |
Wang, C.; Li, B.; Wang, C.; Wu, B. Acta Chim. Sinica 2022, 80, 101. (in Chinese)
doi: 10.6023/A21120564 |
|
(王冲, 李宝林, 王春儒, 吴波, 化学学报, 2022, 80, 101.)
doi: 10.6023/A21120564 |
|
| [22] |
Zhai, Y.; Xu, W.; Meng, X.; Hou, H. Acta Chim. Sinica 2020, 78, 256. (in Chinese)
doi: 10.6023/A19120427 |
|
(翟亚丽, 许文娟, 孟祥茹, 侯红卫, 化学学报, 2020, 78, 256.)
doi: 10.6023/A19120427 |
|
| [23] |
Isac, D. L.; Airinei, A.; Homocianu, M.; Fifere, N.; Cojocaru, C.; Hulubei, C. J. Photoch. Photobio. A. 2020, 390, 112300.
doi: 10.1016/j.jphotochem.2019.112300 |
| [24] |
Sun, J.; Wu, Q.; Weng, W.; Liu, X.; Tan, P.; Sun, L. Acta Chim. Sinica 2020, 78, 1082. (in Chinese)
doi: 10.6023/A20070316 |
|
(孙静静, 吴仇荣, 翁文强, 刘晓勤, 谈朋, 孙林兵, 化学学报, 2020, 78, 1082.)
doi: 10.6023/A20070316 |
|
| [25] |
Zang, Y.; Zou, Q.; Fu, T.; Ng, F.; Fowler, B.; Yang, J.; Li, H.; Steigerwald, M. L.; Nuckolls, C.; Venkataraman, L. Nat. Commun. 2019, 10, 4482.
doi: 10.1038/s41467-019-12487-w |
| [26] |
Huang, X.; Tang, C.; Li, J.; Chen, L.-C.; Zheng, J.; Zhang, P.; Le, J.; Li, R.; Li, X.; Liu, J.; Yang, Y.; Shi, J.; Chen, Z.; Bai, M.; Zhang, H.-L.; Xia, H.; Cheng, J.; Tian, Z.-Q.; Hong, W. Sci. Adv. 2019, 5, eaaw3072.
doi: 10.1126/sciadv.aaw3072 |
| [27] |
Dutta, B. J.; Bhattacharyya, P. K. Int. J. Quantum Chem. 2015, 115, 1459.
doi: 10.1002/qua.24950 |
| [28] |
Shaik, S.; Ramanan, R.; Danovich, D.; Mandal, D. Chem. Soc. Rev. 2018, 47, 5125.
doi: 10.1039/C8CS00354H |
| [29] |
Avdic, I.; Kempfer-Robertson, E. M.; Thompson, L. M. J. Phys. Chem. A 2021, 125, 8238.
doi: 10.1021/acs.jpca.1c06102 pmid: 34494847 |
| [30] |
Yogitha, S. N.; Kumar, B.; Raghavendra; Imranpasha; Gupta, S. K. Mater. Sci. Eng. B 2021, 267, 115094.
doi: 10.1016/j.mseb.2021.115094 |
| [31] |
Alemani, M.; Peters, M. V.; Hecht, S.; Rieder, K.-H.; Moresco, F.; Grill, L. J. Am. Chem. Soc. 2006, 128, 14446.
pmid: 17090013 |
| [32] |
Lu, T.; Chen, Q. ChemPhysChem 2021, 22, 386.
doi: 10.1002/cphc.202000903 |
| [33] |
Meng, L.; Xin, N.; Hu, C.; Wang, J.; Gui, B.; Shi, J.; Wang, C.; Shen, C.; Zhang, G.; Guo, H.; Meng, S.; Guo, X. Nat. Commun. 2019, 10, 1450.
doi: 10.1038/s41467-019-09120-1 |
| [34] |
Stark, J. Nature 1913, 92, 401.
|
| [35] |
Fried, S. D.; Boxer, S. G. Annu. Rev. Biochem. 2017, 86, 387.
doi: 10.1146/annurev-biochem-061516-044432 |
| [36] |
Murgida, D. H.; Hildebrandt, P. Acc. Chem. Res. 2004, 37, 854.
doi: 10.1021/ar0400443 |
| [37] |
Bruot, C.; Hihath, J.; Tao, N. Nat. Nanotechnol. 2012, 7, 35.
doi: 10.1038/nnano.2011.212 |
| [38] |
Chai, J.-D.; Head-Gordon, M. Phys. Chem. Chem. Phys. 2008, 10, 6615.
doi: 10.1039/b810189b |
| [39] |
Hratchian, H. P.; Schlegel, H. B. J. Chem. Phys. 2004, 120, 9918.
pmid: 15268010 |
| [40] |
Lu, T.; Chen, F. J. Comput. Chem. 2012, 33, 580.
doi: 10.1002/jcc.22885 |
| [41] |
Legault, C. Y. CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org.
|
| [42] |
Humphrey, W.; Dalke, A.; Schulten, K. J. Mol. Graph. Model. 1996, 14, 33.
|
| [1] | 黄广龙, 薛小松. “陈试剂”作为三氟甲基源机理的理论研究[J]. 化学学报, 2024, 82(2): 132-137. |
| [2] | 崔国庆, 胡溢玚, 娄颖洁, 周明霞, 李宇明, 王雅君, 姜桂元, 徐春明. CO2加氢制醇类催化剂的设计制备及性能研究进展[J]. 化学学报, 2023, 81(8): 1081-1100. |
| [3] | 付信朴, 王秀玲, 王伟伟, 司锐, 贾春江. 团簇Au/CeO2的制备及其催化CO氧化反应构效关系的研究★[J]. 化学学报, 2023, 81(8): 874-883. |
| [4] | 梁雪峰, 荆剑, 冯昕, 赵勇泽, 唐新员, 何燕, 张立胜, 李慧芳. 共价有机框架COF66/COF366的电子结构: 从单体到二维平面聚合物[J]. 化学学报, 2023, 81(7): 717-724. |
| [5] | 杨磊, 葛娇阳, 王访丽, 吴汪洋, 郑宗祥, 曹洪涛, 王洲, 冉雪芹, 解令海. 一种基于芴的大环结构的有效降低内重组能的理论研究[J]. 化学学报, 2023, 81(6): 613-619. |
| [6] | 张少秦, 李美清, 周中军, 曲泽星. 多共振热激活延迟荧光过程的理论研究[J]. 化学学报, 2023, 81(2): 124-130. |
| [7] | 王娟, 肖华敏, 谢丁, 郭元茹, 潘清江. 铜掺杂与氮化碳复合氧化锌材料结构和二氧化氮气体传感性质的密度泛函理论计算[J]. 化学学报, 2023, 81(11): 1493-1499. |
| [8] | 刘金晶, 杨娜, 李莉, 魏子栋. 铂活性位空间结构调控氧还原机理的理论研究★[J]. 化学学报, 2023, 81(11): 1478-1485. |
| [9] | 栾雪菲, 王聪芝, 夏良树, 石伟群. 铀酰与羧酸和肟基类配体相互作用的理论研究[J]. 化学学报, 2022, 80(6): 708-713. |
| [10] | 郭瑞, 魏星, 曹末云, 张研, 杨云, 樊继斌, 刘剑, 田野, 赵泽坤, 段理. AlAs/InSe范德华异质结构的光学和可调谐电子特性[J]. 化学学报, 2022, 80(4): 526-534. |
| [11] | 杨思明, 刘爱荣, 刘静, 刘钊丽, 张伟贤. 硫化纳米零价铁研究进展: 合成、性质及环境应用[J]. 化学学报, 2022, 80(11): 1536-1554. |
| [12] | 熊昆, 陈伽瑶, 杨娜, 蒋尚坤, 李莉, 魏子栋. 理论探究水溶液条件对TMNxCy催化氮还原性能的增强机制[J]. 化学学报, 2021, 79(9): 1138-1145. |
| [13] | 王英辉, 魏思敏, 段金伟, 王康. 理论研究“受阻路易斯酸碱对”催化的烯醇硅醚氢化反应机理[J]. 化学学报, 2021, 79(9): 1164-1172. |
| [14] | 赵庆如, 蒋茹, 游书力. 铱催化串联不对称烯丙基取代/双键异构化构建轴手性化合物[J]. 化学学报, 2021, 79(9): 1107-1112. |
| [15] | 滑熠龙, 李冬涵, 顾天航, 王伟, 李若繁, 杨建平, 张伟贤. 纳米零价铁富集水溶液中铀的表面化学及应用展望[J]. 化学学报, 2021, 79(8): 1008-1022. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
