化学学报 ›› 2025, Vol. 83 ›› Issue (8): 833-843.DOI: 10.6023/A25030081 上一篇 下一篇
研究论文
成守飞a,b, 李婧c, 凌琳b, 李玉学b,*( ), 吕龙b,*()
), 吕龙b,*()
投稿日期:2025-03-14
发布日期:2025-04-24
通讯作者:
李玉学, 吕龙
基金资助:
Shoufei Chenga,b, Jing Lic, Lin Lingb, Yuxue Lib,*(), Long Lub,*()
Received:2025-03-14
Published:2025-04-24
Contact:
Yuxue Li, Long Lu
Supported by:文章分享

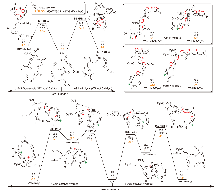
采用M06-2X/6-311+G**方法系统地研究了硝化甘油的分解机理, 包括单分子热分解、无催化水解、酸性杂质(HNO3)催化水解以及金属盐/碱杂质(Zn²+/Mg²+阳离子与OH-/ /Cl-阴离子的组合)参与的分解反应. 此前的理论研究多集中于硝化甘油的单分子分解路径, 而且没有考虑杂质的影响. 本研究发现, 在酸性杂质存在的条件下, 酸催化水解的能垒为28.1 kcal/mol, 比单分子分解的O—NO2均裂能垒降低了5.8 kcal/mol, 是更合理的室温下缓慢分解的机理; 而且水解反应不断释放出HNO3, 使分解速度不断加快; 中强碱Mg(OH)₂和硝化甘油的反应能垒仅为21.3 kcal/mol, 反应速度较快; 虽然该反应是化学计量反应, 但碱作为少量杂质存在时, 也可能触发酸催化分解反应; 酸催化机理可能最接近硝化甘油常温下缓慢分解的真实机理.
成守飞, 李婧, 凌琳, 李玉学, 吕龙. 硝化甘油缓慢分解机理的密度泛函理论研究[J]. 化学学报, 2025, 83(8): 833-843.
Shoufei Cheng, Jing Li, Lin Ling, Yuxue Li, Long Lu. Density Functional Theory Research on Decomposition Mechanisms of Nitroglycerin[J]. Acta Chimica Sinica, 2025, 83(8): 833-843.
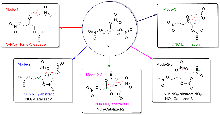

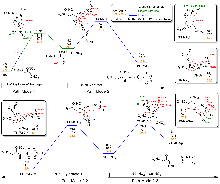



| Pathway | Gas phase | In NG | In H2O | |||||
|---|---|---|---|---|---|---|---|---|
| α | β | α | β | α | β | |||
| Path-Mode-1 | 32.8 | 33.9 | 35.9 | 35.5 | 35.0 | 33.9 | ||
| Path-Mode-2 | 61.7 | 64.0 | 63.6 | 64.4 | 58.9 | 60.3 | ||
| Path-Mode-2-2 | 65.3 | 66.1 | 62.9 | 64.4 | 61.0 | 62.0 | ||
| Path-Mode-2-3 | 67.1 | 65.8 | 67.2 | 66.2 | 64.9 | 64.4 | ||
| Path-Mode-3 | 44.9 | 45.1 | 47.0 | 47.8 | 45.7 | 46.1 | ||
| Pathway | Gas phase | In NG | In H2O | |||||
|---|---|---|---|---|---|---|---|---|
| α | β | α | β | α | β | |||
| Path-Mode-1 | 32.8 | 33.9 | 35.9 | 35.5 | 35.0 | 33.9 | ||
| Path-Mode-2 | 61.7 | 64.0 | 63.6 | 64.4 | 58.9 | 60.3 | ||
| Path-Mode-2-2 | 65.3 | 66.1 | 62.9 | 64.4 | 61.0 | 62.0 | ||
| Path-Mode-2-3 | 67.1 | 65.8 | 67.2 | 66.2 | 64.9 | 64.4 | ||
| Path-Mode-3 | 44.9 | 45.1 | 47.0 | 47.8 | 45.7 | 46.1 | ||





| Pathway | SMD in NG | SMD in H2O | |||
|---|---|---|---|---|---|
| α | β | α | β | ||
| Path-H-Mode-1 | 51.9 | 54.2 | 56.5 | 52.5 | |
| Path-H-Mode-2 | 49.3 | 50.9 | 42.1 | 43.1 | |
| Path-H-Mode-3 | 54.5 | 51.5 | 47.1 | 44.4 | |
| Path-H-Mode-4 | 39.0 | 32.5 | 29.4 | 28.1 | |
| Pathway | SMD in NG | SMD in H2O | |||
|---|---|---|---|---|---|
| α | β | α | β | ||
| Path-H-Mode-1 | 51.9 | 54.2 | 56.5 | 52.5 | |
| Path-H-Mode-2 | 49.3 | 50.9 | 42.1 | 43.1 | |
| Path-H-Mode-3 | 54.5 | 51.5 | 47.1 | 44.4 | |
| Path-H-Mode-4 | 39.0 | 32.5 | 29.4 | 28.1 | |




| M=Zn2+ | M=Mg2+ | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OH- | Cl- | OH- | Cl- | ||||||||||||||
| α | β | α | β | α | β | α | β | α | β | α | β | ||||||
| M-Mode-1 | 47.2 | 44.1 | 45.0 | 44.5 | 45.9 | 42.1 | 43.5 | 38.6 | |||||||||
| M-Mode-2 | 28.7 | 26.3 | 54.1 | 48.3 | 29.2 | 30.1 | 52.6 | 46.1 | |||||||||
| M-Mode-3 | 26.2 | 25.3 | 21.3 | 22.0 | |||||||||||||
| M-Mode-4 | 27.0 | 27.5 | 36.4 | 39.9 | 27.3 | 26.7 | 43.3 | 47.7 | |||||||||
| M=Zn2+ | M=Mg2+ | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OH- | Cl- | OH- | Cl- | ||||||||||||||
| α | β | α | β | α | β | α | β | α | β | α | β | ||||||
| M-Mode-1 | 47.2 | 44.1 | 45.0 | 44.5 | 45.9 | 42.1 | 43.5 | 38.6 | |||||||||
| M-Mode-2 | 28.7 | 26.3 | 54.1 | 48.3 | 29.2 | 30.1 | 52.6 | 46.1 | |||||||||
| M-Mode-3 | 26.2 | 25.3 | 21.3 | 22.0 | |||||||||||||
| M-Mode-4 | 27.0 | 27.5 | 36.4 | 39.9 | 27.3 | 26.7 | 43.3 | 47.7 | |||||||||
| [1] |
|
| [2] |
|
|
(乌尔班斯基著, 火炸药的化学与工艺学II, 牛秉彝, 陈绍亮译, 国防工业出版社, 北京, 1976.)
|
|
| [3] |
|
|
(托马斯•马蒂亚斯著, 张建国, 秦涧译, 高能材料化学, 北京理工大学出版社, 北京, 2016.)
|
|
| [4] |
|
|
(肯尼•范特著, 王康译, 诺贝尔全传, 世界知识出版社, 北京, 2014.)
|
|
| [5] |
|
|
(罗云军, 孙世雄, 高能固体推进剂, 北京理工大学出版社, 北京, 2023.)
|
|
| [6] |
|
| [7] |
|
|
(王振江, 薄月英, 兵工安全技术, 1996, 1, 37.)
|
|
| [8] |
|
|
(张彦才, 江业梁, 工业安全与防尘, 1990, 6, 37.)
|
|
| [9] |
|
|
(储绍华, 现代兵器, 1983, 6, 42.)
|
|
| [10] |
|
|
(张国顺, 王泽溥, 火炸药及其制品燃烧爆炸事故及其预防措施, 兵器工业出版社, 北京, 2009.)
|
|
| [11] |
|
|
(杨根, 彭松, 池旭辉, 固体火箭技术, 2009, 32, 650.)
|
|
| [12] |
|
| [13] |
|
|
(何彬, 卢先明, 孟子晖, 兵器装备工程学报, 2022, 43, 108.)
|
|
| [14] |
|
| [15] |
|
|
(姚子云, 罗秉和, 中北大学学报(自然科学版), 1989, 2, 42.)
|
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
|
(裴海潮, 吴婉娥, 付潇, 固体火箭技术, 2016, 39, 538.)
|
|
| [20] |
|
| [21] |
|
| [22] |
|
|
(裴立冠, 董可海, 唐岩辉, 含能材料, 2017, 25, 804.)
|
|
| [23] |
|
| [24] |
|
|
(丁超, 吴婉娥, 裴海潮, 化学推进剂与高分子材料, 2017, 15, 4.)
|
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
|
(刘子茹, 含能材料热分析, 国防工业出版社, 北京, 2008.)
|
|
| [29] |
|
|
(刘艳, 刘子如, 邱刚, 阴翠梅, 火炸药学报, 2001, 24, 26.)
|
|
| [30] |
|
| [31] |
|
|
(安晋川, 硝化甘油, 火炸药丛书, 1974.)
|
|
| [32] |
|
|
(方维吾, 夏正寅, 武超宇, 硝化甘油, 常规兵器工业安全技术事故资料丛书, 国防工业出版社, 北京, 1984.)
|
|
| [33] |
|
|
(曲开社, 系统工程理论与实践, 1996, 16, 82.)
|
|
| [34] |
|
| [35] |
See supporting information.
|
| [36] |
|
|
(凌琳, 王健, 李婧, 李玉学, 吕龙, 有机化学, 2023, 43, 285.)
doi: 10.6023/cjoc202206027 |
|
| [37] |
|
|
(Dean, J. A.主编, 兰氏化学手册, 尚久方译, 科学出版社, 北京, 1991.)
|
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
doi: 10.6023/A23020056 |
|
(杨洁, 凌琳, 李玉学, 吕龙, 化学学报, 2023, 81, 328.)
doi: 10.6023/A23020056 |
|
| [46] |
|
|
(崔勇康, 成守飞, 凌琳, 李玉学, 吕龙, 化学学报, 2024, 82, 377.)
doi: 10.6023/A24010017 |
|
| [47] |
|
| [1] | 王鑫, 史燚威, 杨瑞杰, 宋志国, 王敏. 含苯磺酸类配体的单核Cd(II)配合物催化无溶剂“一锅法”Biginelli反应[J]. 化学学报, 2025, 83(7): 674-684. |
| [2] | 马玲玲, 凌琳, 吴亚明, 李玉学, 吕龙. 1,3,5-三乙氧基-2,4,6-三硝基苯的连续化制备反应机理的理论研究[J]. 化学学报, 2025, 83(4): 360-368. |
| [3] | 卢一林, 董盛杰, 崔方超, 薄婷婷, 毛卓. 希托夫紫磷烯/SnS2范德华异质结作为直接全解水光催化剂的理论构建[J]. 化学学报, 2025, 83(4): 377-389. |
| [4] | 陈铭晖, 张博心, 魏滔, 孙兆雪, 冯亚青, 张宝. 三嗪共价骨架材料的层间位错行为及其光生载流子动力学理论研究[J]. 化学学报, 2025, 83(2): 93-100. |
| [5] | 马莹, 陈维希, 刘羽辰, 刘子义, 吴涛, 陆安慧, 王东琪. 六方氮化硼氧化模式的密度泛函理论研究[J]. 化学学报, 2025, 83(1): 52-59. |
| [6] | 王治业, 肖博怀. 利用平面σ-芳香性增强电子输运能力[J]. 化学学报, 2024, 82(5): 520-526. |
| [7] | 赵雨晴, 梁栋, 贾吉慧, 余荣民, 卢灿忠. 具有双吸电子基团D-A型配体的Ag(I)发光配合物的合成与性能研究[J]. 化学学报, 2024, 82(5): 486-492. |
| [8] | 崔勇康, 成守飞, 凌琳, 李玉学, 吕龙. 二氟氨基二硝甲基芳香杂环含能材料的理论研究[J]. 化学学报, 2024, 82(4): 377-386. |
| [9] | 赵玉强, 张霞, 杨芸如, 朱立平, 周莹. 聚集诱导发射光笼分子的设计合成及原位光激活成像研究[J]. 化学学报, 2024, 82(3): 265-273. |
| [10] | 黄广龙, 薛小松. “陈试剂”作为三氟甲基源机理的理论研究[J]. 化学学报, 2024, 82(2): 132-137. |
| [11] | 黄伊晨, 聂长明, 王聪芝, 陈树森, 宋艳, 李昊, 石伟群. 羟基和氨基取代偕胺肟用于海水提铀的理论研究[J]. 化学学报, 2024, 82(10): 1050-1057. |
| [12] | 梁雪峰, 荆剑, 冯昕, 赵勇泽, 唐新员, 何燕, 张立胜, 李慧芳. 共价有机框架COF66/COF366的电子结构: 从单体到二维平面聚合物[J]. 化学学报, 2023, 81(7): 717-724. |
| [13] | 杨磊, 葛娇阳, 王访丽, 吴汪洋, 郑宗祥, 曹洪涛, 王洲, 冉雪芹, 解令海. 一种基于芴的大环结构的有效降低内重组能的理论研究[J]. 化学学报, 2023, 81(6): 613-619. |
| [14] | 杨洁, 凌琳, 李玉学, 吕龙. 高氯酸铵热分解机理的密度泛函理论研究[J]. 化学学报, 2023, 81(4): 328-337. |
| [15] | 张少秦, 李美清, 周中军, 曲泽星. 多共振热激活延迟荧光过程的理论研究[J]. 化学学报, 2023, 81(2): 124-130. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||
