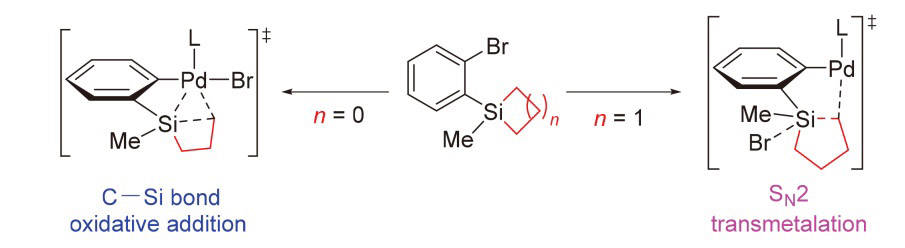
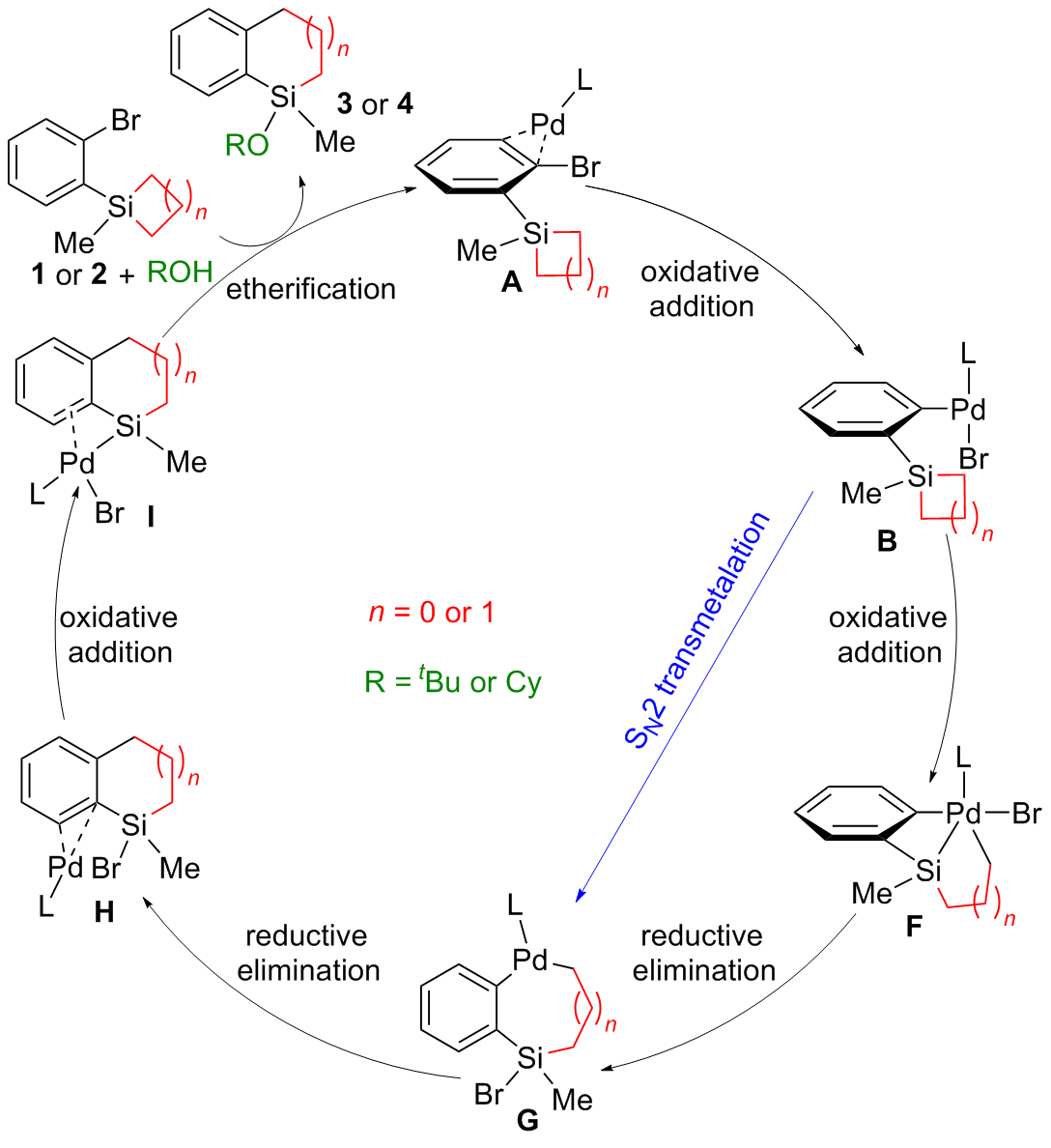
| [1] |
Ishida, N.; Okumura, S.; Kawasaki, T.; Murakami, M. Angew. Chem., Int. Ed. 2018, 57, 11399.
|
| [2] |
Wu, Y.; Wang, P. Angew. Chem., Int. Ed. 2022, 61, e202205382.
|
| [3] |
Wang, Q.; Jiang, H.-M.; Zhuo, S.; Xu, L.-P. Catal. Sci. Technol. 2022, 12, 135.
|
| [4] |
Rémond, E.; Martin, C.; Martinez, J.; Cavelier, F. Chem. Rev. 2016, 116, 11654.
|
| [5] |
Hailes, R. L. N.; Oliver, A. M.; Gwyther, J.; Whittell, G. R.; Manners, I. Chem. Soc. Rev. 2016, 45, 5358.
|
| [6] |
Su, T. A.; Li, H.; Klausen, R. S.; Kim, N. T.; Neupane, M.; Leighton, J. L.; Steigerwald, M. L.; Venkataraman, L.; Nuckolls, C. Acc. Chem. Res. 2017, 50, 1088.
|
| [7] |
Gan, W.-E.; Wu, Y.-S.; Wu, B.; Fang, C.-Y.; Cao, J.; Xu, Z.; Xu, L.-W. Angew. Chem., Int. Ed. 2024, 63, e202317973.
|
| [8] |
Wu, L.; Zhang, L.; Guo, J.; Gao, J.; Ding, Y.; Ke, J.; He, C. Angew. Chem., Int. Ed. 2024, 63, e202413753.
|
| [9] |
Cui, D.; Feng, Y.; Gan, Y.; Yin, J.; Wang, W.; Fan, Y.; Gao, L.; Ke, B.; Song, Z. Org. Chem. Front. 2022, 9, 5453.
|
| [10] |
Luo, G.; Chen, L.; Li, Y.; Fan, Y.; Wang, D.; Yang, Y.; Gao, L.; Jiang, R.; Song, Z. Org. Chem. Front. 2021, 8, 5941.
|
| [11] |
Franz, A. K.; Wilson, S. O. J. Med. Chem., 2013, 56, 388.
|
| [12] |
Ramesh, R.; Reddy, D. S. J. Med. Chem, 2017, 61, 3779.
|
| [13] |
Liu, Y.-J.; Liu, Y.-H.; Zhang, Z.-Z.; Yan, S.-Y.; Chen, K.; Shi, B.-F. Angew. Chem., Int. Ed. 2016, 55, 13859.
|
| [14] |
Wang, W.; Zhou, S.; Li, L.; He, Y.; Dong, X.; Gao, L.; Wang, Q.; Song, Z. J. Am. Chem. Soc. 2021, 143, 11141.
|
| [15] |
Kinnaird, J. W. A.; Ng, P. Y.; Kubota, K.; Wang, X.; Leighton, J. L. J. Am. Chem. Soc. 2002, 124, 7920.
pmid: 12095334
|
| [16] |
Zhang, X.; Houk, K. N.; Leighton, J. L. Angew. Chem., Int. Ed. 2005, 44, 938.
|
| [17] |
Li, L.; Zhang, Y.; Gao, L.; Song, Z. Tetrahedron Lett. 2015, 56, 1466.
|
| [18] |
Zhao, W. T.; Gao, F.; Zhao, D. Angew. Chem. Int. Ed. 2018, 57, 6329.
|
| [19] |
Chen, H.; Peng, J.; Pang, Q.; Du, H.; Huang, L.; Gao, L.; Lan, Y.; Yang, C.; Song, Z. Angew. Chem., Int. Ed. 2022, 61, e202212889.
|
| [20] |
Chen, H.; Zhang, H.; Du, H.; Kuang, Y.; Pang, Q.; Wang, W.; Yang, C.; Song, Z. Org. Lett. 2023, 25, 1558.
|
| [21] |
Tang, X.; Zhang, Y.; Tang, Y.; Li, Y.; Zhou, J.; Wang, D.; Gao, L.; Su, Z.; Song, Z. ACS Catal. 2022, 12, 5185.
|
| [22] |
Zhu, W.; Xu, L. Chin. J. Org. Chem. 2023, 43, 362 (in Chinese).
|
|
(祝炜轲, 徐利文, 有机化学, 2023, 43, 362.)
doi: 10.6023/cjoc202300003
|
| [23] |
Yin, G.; Xu, L. Chin. J. Org. Chem. 2021, 41, 4839 (in Chinese).
|
|
(尹官武, 徐利文, 有机化学, 2021, 41, 4839.)
doi: 10.6023/cjoc202100094
|
| [24] |
Chen, S.; He, X.; Chen, J.; Zhang, W.; Yang, Y.; Liu, S., Lan, Y.; Houk, K. N.; Shen, X. Angew. Chem., Int. Ed. 2022, 61, e202213431.
|
| [25] |
Tang, X.; Tang, Y.; Peng, J.; Du, H.; Huang, L.; Gao, J.; Liu, S.; Wang, D.; Wang, W.; Gao, L.; Lan, Y.; Song, Z. J. Am. Chem. Soc. 2024, 146, 26639.
|
| [26] |
Huo, J.; Zhong, K.; Xue, Y.; Lyu, M. M.; Ping, Y.; Liu, Z.; Lan, Y.; Wang, J. J. Am. Chem. Soc. 2021, 143, 12968.
|
| [27] |
Qin, Y.; Han, J.-L.; Ju, C.-W.; Zhao, D. Angew. Chem., Int. Ed. 2020, 59, 8481.
|
| [28] |
Chen, W. J.; Lin, Z. Dalton Trans. 2014, 43, 11138.
|
| [29] |
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019.
|
| [30] |
Becke, A. D. Phys. Rev. A 1988, 38, 3098.
|
| [31] |
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
doi: 10.1103/physrevb.37.785
pmid: 9944570
|
| [32] |
Becke, A. D. J. Chem. Phys. 1993, 98, 1372.
|
| [33] |
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104.
|
| [34] |
Becke, A. D.; Johnson, E. R. J. Chem. Phys. 2005, 123, 154101.
|
| [35] |
Johnson, E. R.; Becke, A. D. J. Chem. Phys. 2006, 124, 174104.
|
| [36] |
Fukui, K. Acc. Chem. Res. 1981, 14, 363.
|
| [37] |
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378.
|
| [38] |
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215.
|
 ), 邓远程a, 李嘉瑜a, 郭益豪a, 王广凤a, 段阿冰c,*(
), 邓远程a, 李嘉瑜a, 郭益豪a, 王广凤a, 段阿冰c,*(