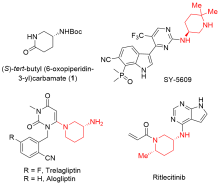
| [1] |
Vitaku, E. V.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257.
|
| [2] |
Abdelshaheed, M. M.; Fawzy, I. M.; El-Subbagh, H. I.; Youssef, K. M. Future J. Pharm. Sci. 2021, 7, 188.
|
| [3] |
Chen, Q.-S.; Li, J.-Q.; Zhang, Q.-W. Pharm. Fronts 2023, 5, e1.
|
| [4] |
Frolov, N.; Vereshchagin, A. N. Int. J. Mol. Sci. 2023, 24, 2937.
|
| [5] |
Dowarah, J.; Singh, V. P. Bioorg. Med. Chem. 2020, 28, 115263.
|
| [6] |
Goel, P.; Alam, O.; Naim, M. J.; Nawaz, F.; Iqbal, M.; Alam, M. I. Eur. J. Med. Chem. 2018, 157, 480.
|
| [7] |
Rkash, R.; Asati, V.; Kashaw, S. K.; Agarwal, S.; Parwani, D.; Bhattacharya, S.; Mallick, C. Mini-Rev. Med. Chem. 2021, 21, 362.
|
| [8] |
Sun, W.; Liao, A.; Lei, L.; Tang, X.; Wang, Y.; Wu, J. Chin. Chem. Lett. 2025, 36, 109855.
|
| [9] |
Marineau, J. J.; Hamman, K. B.; Hu, S.; Alnemy, S.; Mihalich, J.; Kabro, A.; Whitmore, K. M.; Winter, D. K.; Roy, S.; Ciblat, S.; Ke, N.; Savinainen, A.; Wilsily, A.; Malojcic, G.; Zahler, R.; Schmidt, D.; Bradley, M. J.; Waters, N. J.; Chuaqui, C. J. Med. Chem. 2022, 65, 1458.
|
| [10] |
Johannessen, L.; Henry, S.; Sawant, P.; D’lppolito, A.; Ke, N.; Lefkowitz, A.; Eaton, M.; Dworakowski, W.; Rosario, M.; Hodgson, G. Blood 2021, 138, 1205.
|
| [11] |
Blair, H. A. Drugs 2023, 83, 1315.
|
| [12] |
Liu, J.; Solan, R.; Wolk, R.; Plotka, A.; O'Gorman, M. T.; Winton, J. A.; Kaplan, J.; Purohit, V. S. Br. J. Clin. Pharmacol. 2023, 89, 2208.
|
| [13] |
Zhang, Z.; Wallace, M. B.; Feng, J.; Stafford, J. A.; Skene, R. J.; Shi, L.; Lee, B.; Aertgeerts, K.; Jennings, A.; Rongda, A.; Xu, R.; Kassel, D. B.; Kaldor, S. W.; Navre, M.; Webb, D. R.; Gwaltney II, S. L. J. Med. Chem. 2011, 54, 510.
|
| [14] |
Grimshaw, C. E.; Jennings, A.; Kamran, R.; Ueno, H.; Nishigaki, N.; Kossaka, T.; Tani, A.; Sano, H.; Kinugawa, Y.; Koumura, E.; Shi, L.; Takeuchi, K. PloS One 2016, 11, e0157509.
|
| [15] |
Feng, J.; Zhang, Z.; Wallace, M. B.; Stafford, J. A.; Kaldor, S. W.; Kassel, D. B.; Navre, M.; Shi, L.; Skene, R. J.; Asakawa, T.; Takeuchi, K.; Xu, R.; Webb, D. R.; Gwaltney, S. L. J. Med. Chem. 2007, 50, 2297.
|
| [16] |
Fujita, H.; Taniai, H.; Murayama, H.; Ohshiro, H.; Hayashi, H.; Sato, S.; Kikuchi, N.; Komatsu, T.; Komatsu, K.; Komatsu, K.; Narita, T.; Yamada, Y. Endocr. J. 2014, 61, 159.
|
| [17] |
Panday, S. K.; Langlois, N. Tetrahedron Lett. 1995, 36, 8205.
|
| [18] |
Langlois, N.; Van Bac, N.; Dahuron, N.; Delcroix, J.-M.; Deyine, A.; Griffart-Bruned, D.; Chiaroni, A.; Riche, C. Tetrahedron 1995, 51, 3571.
|
| [19] |
Langlois, N. Tetrahedron Lett. 2002, 43, 9531.
|
| [20] |
Masse, J.; Langlois, N. Heterocycles 2009, 77, 417.
|
| [21] |
Thomas, A.; Bhavar, P. K.; Lingam, V. S. P. R.; Joshi, N. K. WO 2004/022536, 2004.
|
| [22] |
Albers, R.; Ayala, L.; Clareen, S. S.; Delgado Mederos, M. M.; Hilgraf, R.; Hedge, S.; Hughes, K.; Kois, A.; Plantevin-Krenitsky, V.; Mccarrick, M.; Nadolny, L.; Palanki, M.; Sahasrabudhe, K.; Sapienza, J.; Satoh, Y.; Sloss, M.; Sudbeck, E.; Wright, J. WO 2006/076595, 2006.
|
| [23] |
Do, S.; Hu, H.; Kolesnikov, A.; Tsui, V. H.; Wang, X. WO 2014/001377, 2014.
|
| [24] |
Marineau, J. J.; Chuaqui, C.; Ciblat, S.; Kabro, A.; Piras, H.; Whitmore, K. M.; Lund, K.-L. WO 2019/143719, 2019.
|
| [25] |
Marineau, J. J.; Chuaqui, C. WO 2020/093011, 2020.
|
| [26] |
Li, L.; Zhang, Y. CN 104098563, 2014.
|
| [27] |
Huang, S.-B.; Nelson, J. S.; Weller, D. D. J. Org. Chem. 1991, 56, 6007.
|
| [28] |
Ghosh, A. K.; Leshchenko-Yashchuk, S.; Anderson, D. D.; Bal- dridge, A.; Noetzel, M.; Miller, H. B.; Tie, Y.; Wang, Y.-F.; Koh, Y.; Weber, I. T.; Mitsuya, H. J. Med. Chem. 2009, 52, 3902.
|
| [29] |
Langlois, N. Org. Lett. 2002, 4, 185.
|
| [30] |
Tan, H.; Reed, M.; Gahm, K. H.; King, T.; Seran, M. D.; Bostick, T.; Luu, V.; Semin, D.; Cheetham, J.; Larsen, R.; Martinelli, M.; Reider, P. Org. Process Res. Dev. 2008, 12, 58.
|
 ), 杨鹏a,*(
), 杨鹏a,*(